



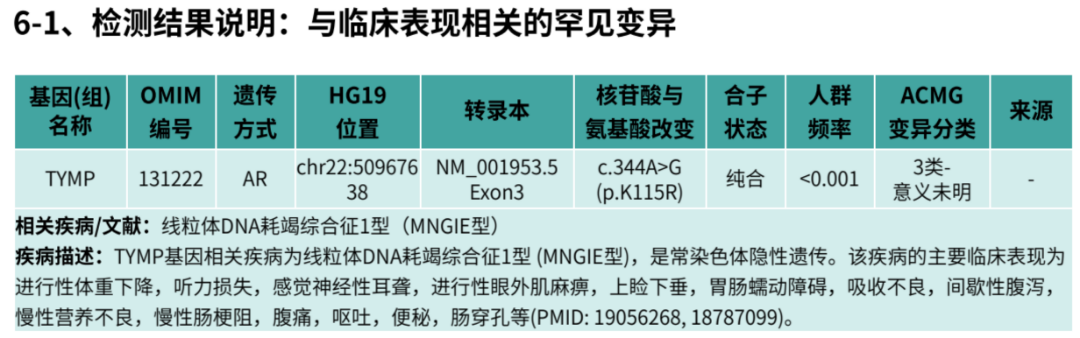
200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:中南大学湘雅三医院消化内科 聂凯 艾飞艳 王晓艳
PART.01
病例介绍
第一次就诊:被忽视的腹胀信号(2013-2019)
患者马某某,女性,55岁,湖南湘西人。2013年初无明显诱因出现反复腹胀,初起症状隐匿,未予重视。2015年腹胀加重,伴餐后早饱、嗳气,当地医院行相关检查考虑消化不良,予促动力药及消化酶治疗后症状稍有缓解,但腹胀仍反复发作。2年前受凉后出现腹泻,大便2~3次/天,为糊状便,伴腹胀、反酸嗳气、乏力、头晕,无黏液脓血便、黑便便血,无腹痛、恶心呕吐、里急后重等不适,至当地医院就诊,大便OB阳性、中度贫血,胃镜示:胆汁反流性胃炎,肠镜未见异常。予以护胃、止血、补充造血原料等对症支持治疗,好转出院。患者自幼体形消瘦,家族中父母及祖父母均为近亲结婚(表兄妹),其姐亦有类似消瘦体型。这一重要的遗传背景在当时未被充分重视,为后续诊断埋下伏笔。
病例特点初析: 中年女性,慢性腹胀,初始症状不典型,按功能性消化不良治疗效果不佳。家族史提示近亲婚配,但缺乏特异性指向。
第二次就诊:消化道出血敲响警钟(2019-2020)
2019年12月,患者因“腹泻2年,大便隐血持续阳性”首次就诊于我院中医科。腹泻表现为每日2~3次糊状便,无黏液脓血,伴乏力、头晕。查血红蛋白76g/L,大便OB(+)。小肠CTE显示:回肠第2~3段肠壁稍均匀增厚,强化扫描黏膜面明显强化,邻近肠系膜多发肿大淋巴结(大者短径约0.9cm);十二指肠降段憩室。T.SPOT-TB(+),但结核抗体、PPD皮试均阴性。
当时诊断困境: 小肠CTE虽提示炎性改变,但缺乏典型克罗恩病(CD)的纵行溃疡、铺路石样改变;结核筛查呈矛盾结果;患者拒绝进一步行胶囊内镜或小肠镜检查。经艾灸、护胃及补充造血原料等对症治疗,症状部分缓解后出院,未能明确诊断。
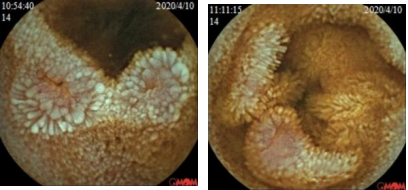
图1 胶囊内镜(胶囊运行4小时23分至12小时1分,可见散在多个片状、横行或纵行糜烂)
转折点: 2020年4月再次因腹胀腹泻就诊于当地医院,完善大便OB阳性,胶囊内镜(胶囊运行4小时23分至12小时1分,可见散在多个片状、横行或纵行糜烂),内镜诊断:1.小肠炎 2.小肠血管显露 3.小肠憩室。入院后予以输血、调节肠道菌群、补铁等对症支持治疗。出院诊断:1.小肠病变性质待定,克罗恩病待排,2.缺铁性贫血,3.胆汁反流性胃炎,4.低蛋白血症。2020年10月,患者因同样原因再次入住当地中医院,查大便OB仍阳性,贫血持续存在。此时,消化科医生首次提出“小肠疾病导致慢性失血”的假设,但仍未跳出“炎症性肠病”的思维定式。
第三次就诊:MDT陷入诊断僵局(2021年7月)
2021年7月,患者第一次入住我科。入院查体:营养不良,贫血貌,BMI 16.2kg/m²。实验室检查:Hb 68 g/L,白蛋白28.5 g/L,CRP 55.92 mg/L,ESR 39 mm/h。三次大便培养、CMV、艰难梭菌、免疫全套均阴性。ANA 1:160阳性(颗粒型),EB-DNA 1.171E+03拷贝/ml。小肠CTE复查显示回肠病变同前,未见进展。
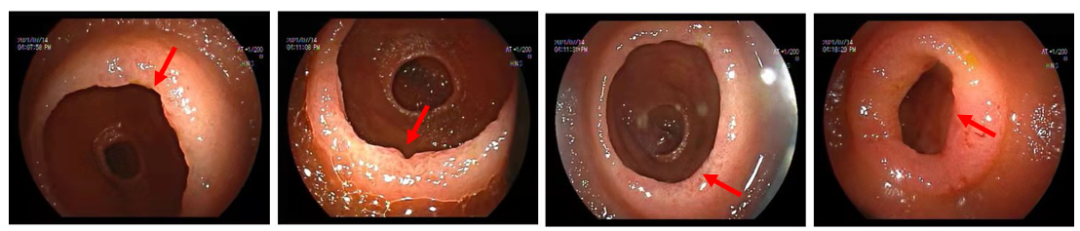
图2 经肛小肠镜检查发现空肠远端及近段回肠多发环形狭窄,散在糜烂及不规则溃疡,溃疡最大径约0.8 cm,呈横向分布。
关键检查揭晓: 经肛小肠镜检查发现空肠远端及近段回肠多发环形狭窄,散在糜烂及不规则溃疡,溃疡最大径约0.8 cm,呈横向分布。活检病理示:检材零碎的小肠黏膜,局灶绒毛稍圆钝及隐窝延长,偶见分支,间质水肿,未见明显肉芽肿及非干酪样坏死。
MDT首次讨论:首次讨论倾向于隐源性多灶性溃疡性狭窄性小肠炎(CMUSE)。消化内科、影像科、病理科联合讨论认为,病变呈节段性分布、以狭窄为主、病理缺乏典型CD表现,倾向于“隐源性多灶性溃疡性狭窄性小肠炎(CMUSE)”。然而,CMUSE多为中年发病,且对激素反应良好,与患者55岁发病、病程迁延8年不符。诊断仍存疑虑,建议密切随访。
第四次就诊:发热危机与诊断突破(2021年9月)
2021年9月3日,患者因“发热半月”再次入院。体温最高达40℃,伴咳嗽、咳痰。查体:双肺呼吸音粗,右肺少许湿啰音。血常规:WBC 8.2×109/L,RBC 2.63× 1012/L,Hb降至68 g/L,PCT 0.31 ng/ml。胸部CT示双肺炎症,心包少量积液。
感染筛查迷局: 全血培养阴性,痰培养示大肠埃希菌及热带假丝酵母菌。考虑真菌为机会性感染,先经验性予哌拉西林他唑巴坦抗感染后体温恢复正常,但腹胀、腹泻无改善,消化道症状与感染不同步。
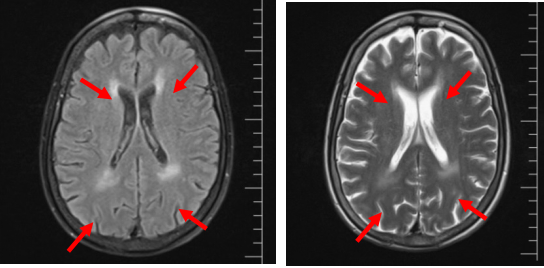
神经科会诊意外发现:神经内科会诊时详细追问病史,患者诉近年出现进行性听力下降、手足麻木,查体发现腱反射减弱。神经肌电图提示:多发慢性周围神经病,运动与感觉神经均有受累,以下肢为主;脱髓鞘伴轴索损害。头颅MRI示脑白质病变。
图3 头部MRI提示脑白质病变
诊断线索串联: 消化症状(腹泻、腹胀、慢性失血)+恶病质(BMI 16.2)+周围神经病变+听力下降+脑白质病变+近亲婚配家族史,高度提示线粒体神经胃肠型脑肌病(MNGIE)。
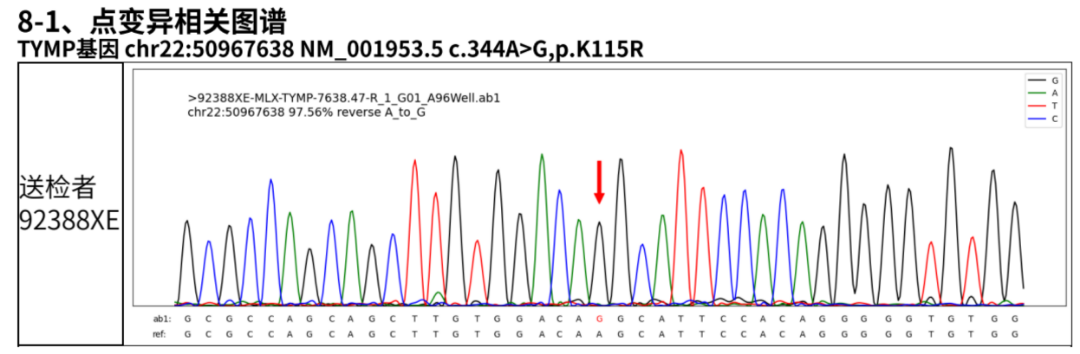
图4 基因检测确诊: 外周血全外显子测序发现TYMP基因c.866A>G(p.His289Arg)纯合突变,其父母均为杂合携带者。
基因检测确诊: 外周血全外显子测序发现TYMP基因c.866A>G(p.His289Arg)纯合突变,其父母均为杂合携带者。血浆脱氧胸苷(dThd)水平检测达8.6μmol/L(正常<0.05),脱氧尿苷(dUrd)12.3 μmol/L(正常<0.05)。白细胞胸苷磷酸化酶(TP)活性检测<5%正常值。最终确诊为线粒体神经胃肠型脑肌病(MNGIE)。
PART.02
诊疗提升
问题一:为何MNGIE如此易误诊为炎性肠病?

MNGIE与CD/UC在消化道表现上有显著重叠:两者均可表现为腹痛、腹泻、肠壁增厚、溃疡形成。
但MNGIE有其独特特征,包括以下4个方面。
1. 疾病谱更广:除肠道症状外,常合并眼外肌麻痹、周围神经病、听力下降、脑白质病变等多系统损害。
2. 病理特征不同:MNGIE肠黏膜活检表现为绒毛钝缩、隐窝延长,但无肉芽肿、无裂隙样溃疡。
3. 影像学差异:MNGIE的小肠病变以均匀增厚为主,强化明显,但狭窄相对均匀,瘘管、脓肿少见。
4. 家族史关键:本例患者父母近亲婚配是重要诊断线索,CD则多为散发。

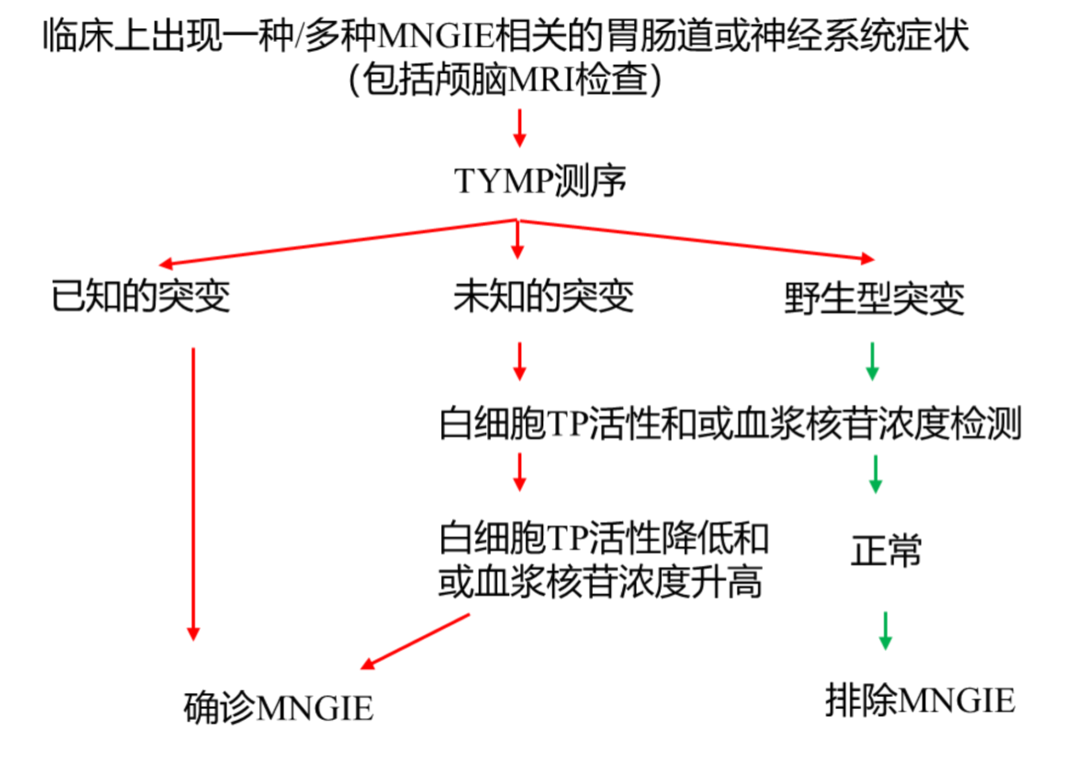
问题2:如何把握基因检测的时机与指征?
本病例从首次就诊到确诊历时8年,关键转折在于MDT时主动提出基因检测。我们建议对以下“预警红线”患者应尽早行基因筛查:
• 小肠疾病+神经系统症状(周围神经病、眼肌麻痹、听力障碍);
• 不明原因消瘦(BMI<18)+慢性消化道症状;
• 近亲婚配家族史+早发消化系统疾病;
• 常规IBD治疗无效或反应不佳;
• 头颅MRI提示脑白质病变。
问题3:MNGIE的阶梯式诊断流程如何建立?
参考国际MNGIE诊断标准,推荐以下流程:
第一阶段(临床筛查):同时存在胃肠动力障碍、恶病质、神经系统症状三联征;
第二阶段(实验室筛查):血浆dThd>3 μmol/L或dUrd>5 μmol/L;
第三阶段(功能检测):白细胞TP酶活性降低;
第四阶段(基因确诊):TYMP基因纯合或复合杂合突变;
本病例符合全部四阶段标准,诊断等级为确诊。
背景知识补充
MNGIE:被低估的罕见病
MNGIE是一种常染色体隐性遗传病,由TYMP基因突变导致胸苷磷酸化酶(TP)活性缺失,造成血浆脱氧核苷蓄积,进而损害线粒体DNA复制。全球报道不足200例,患病率约1/1,000,000。
临床分型与预后:
• 早发型(<40岁):病情进展迅速,平均生存期35-37岁
• 晚发型(>40岁):症状较轻,本例55岁发病属于罕见晚发型
• 存活率:>50岁患者5年存活率仅5%,本例确诊时已属疾病晚期
消化内科医师必须警惕的特征:
1. 假性肠梗阻:约57%患者以胃肠道症状首发,表现为腹胀、呕吐,但肠镜常无明显异常
2. 吸收不良:由于肠壁平滑肌线粒体功能障碍,导致营养吸收障碍,表现为低蛋白血症、贫血
3. 对IBD治疗无反应:激素、免疫抑制剂治疗无效是本病的"反向指征"
各科室专家点评
消化内科点评:
本病例最深刻的教训在于,当我们面对“不典型IBD”时,应建立“诊疗时钟”概念——若规范治疗3个月无效,必须重新审视诊断。MNGIE与IBD的核心区别在于多系统受累,特别是神经系统症状的隐匿性出现。建议对所有慢性小肠疾病患者常规询问听力、视力变化及家族史。
神经内科点评:
患者周围神经病变以轴索损害为主,伴脱髓鞘改变,这是MNGIE的典型电生理特征。我们遇到不明原因的慢性进行性眼外肌麻痹或周围神经病时,也应主动追问消化道症状。跨科室的'症状串联'是诊断罕见病的关键。
医学遗传科点评:
近亲婚配家族史是本病例的诊断突破口。TYMP基因c.866A>G突变在中国南方人群中携带率约0.3%,近亲婚配使纯合子概率提升至25%。对于基层医院,当遇到家族聚集的慢性消耗性疾病,应首选家系图谱分析,再决定基因检测范围。
营养科点评:
患者入院时NRS-2002评分6分,重度营养不良。MNGIE的营养支持有其特殊性:需避免过度喂养加重代谢负担,推荐中等热量密度25 kcal/(kg·d)、高蛋白1.5 g/(kg·d)的肠内营养配方。本例患者经3个月营养干预,白蛋白从28.5 g/L升至35.2 g/L,为后续治疗奠定基础。
经验总结与临床启示
诊断思维的“三线突破”原则:
• 一线:常见疾病(IBD)→规范治疗无效时
• 二线:少见疾病(CMUSE、肠结核、血管炎)→MDT仍不能解释时
• 三线:罕见病(MNGIE、先天性失氯腹泻)→必须寻找“red flag”(红旗征)
02
家族史的“三维挖掘”:
• 深度:追问三代以内近亲婚配史
• 宽度:询问所有血缘亲属的相似症状
• 高度:绘制家系遗传图谱,标注患者表型
03
多学科协作的“时间窗口”:
本病例从首次MDT到确诊历时2个月,关键在于第二次MDT时主动邀请神经内科参与。建议对复杂小肠疾病,MDT应常规包含神经科、遗传科、营养科,实现"一站式"评估。
04
罕见病诊断的“5年生存率”警示:
MNGIE虽罕见,但一旦漏诊,患者将失去接受造血干细胞移植的机会。消化科医生应成为罕见病的“守门员”,当遇到“IBD不典型”时,要敢于怀疑、勇于求证。
最终诊断
01
线粒体神经胃肠型脑肌病(MNGIE,晚发型)
02
慢性中度失血性贫血(缺铁性贫血)
03
低蛋白血症(重度营养不良)
04
双肺肺炎(细菌感染)
05
周围神经病变(轴索损害伴脱髓鞘)
专家小结
中南大学湘雅三医院消化内科王晓艳教授点评:
本病例的“一波三折”本质上是罕见病诊断困难的缩影。第一次曲折在于将MNGIE误认为功能性消化不良;第二次曲折在于CTE表现误导为IBD;第三次曲折在于发热感染掩盖了原发病;第四次曲折才通过MDT和基因检测揭晓真相。整个过程历时8年,给患者及家庭带来巨大负担。
四大启示
1. 建立“诊断时钟”:对不明原因消化道症状,若2年内不能确诊,必须启动罕见病筛查流程
2. 强化“系统思维”:消化道症状永远不是孤立的,必须主动寻找肠外表现
3. 重视“遗传背景”:在边远贫困地区,近亲婚配仍较常见,这是诊断常染色体隐性遗传病的重要线索
4. 推动“技术下沉”:血浆dThd/dUrd检测并非常规项目,建议在有条件的中心推广,建立区域检测网络
本病例的成功诊断,体现了MDT在罕见病诊治中的核心价值。未来,我们应建立MNGIE患者登记制度,开展多中心临床研究,为更多患者带来生的希望。

湘雅三医院消化内科主任、国家临床重点专科学科带头人、二级教授、博导
“湖南省医学领军人才”“湖南省创新群体带头人”
中华医学会消化病学分会 委员
中国医师协会消化医师分会 常务委员
湖南省医学会消化病学委员会 主任委员
中华医学会消化内镜学分会大数据协作组 副组长
中华医学会消化病学分会IBD学组 委员
湖南省炎症与肿瘤重点实验室 主任
湖南省消化内镜微创诊治临床医学研究中心 主任
湖南省医学会超声内镜学组 组长
主要从事消化系统疾病内镜诊治、消化系统炎症与肿瘤的基础及临床研究。擅长EUS、ESD及ERCP等内镜诊治技术。主持国家自然科学基金区域重点项目及面上项目7项,以通讯作者在Cell metabolism、Nature Communications、EClinicalMedicine、Diabetes Care等期刊发表SCI论文近100篇,IF>10分的18篇。获湖南省科技进步奖及教学奖5项,获国家专利7项,转化1项。

中南大学湘雅三医院消化内科博士,副主任医师
中国医师协会内镜医师分会小肠内镜学组委员
湖南省康复医学会消化病康复专业委员会肠道微生态学组副组长
湖南省女医师协会肠道微生态健康专委会常委
湖南省女医师协会消化专委会委员
湖南省健康管理学会疑难肠病专委会委员
湖南省小肠疾病诊疗联盟秘书
湖南省IBD医师联盟成员

博士,湘雅三医院消化内科主治医师
中华医学会消化病学分会微生态学组委员
中华医学会消化病学分会流行病学协作组委员
消化病学分会IBD学组青年俱乐部成员
欧洲炎症性肠病组织壁报及基金评审
韩国肠病协会杰出研究者奖得主
KASID青年研究奖得主
亚洲炎症性肠病组织杰出壁报奖得主
2024年IBD-PicShow大赛第一名
全国消化年会优秀论文奖
ESI 高被引和热点论文作者
主要从事消化道疾病及内镜下诊疗、炎症性肠病的基础及临床研究。主持国自然青年基金和省自然青年基金各1项,参与国家自然科学基金4项,一作和通讯在Gastroenterology等杂志发表论文。担任杂志JCC Plus编委、SCI杂志Science advance, Clinical Infectious Diseases、Inflammatory bowel diseases,等SCI杂志审稿人。受邀在亚洲炎症性肠病组织年会、韩国肠病国际会议、全国消化年会上口头交流,并在DDW、ECCO、APDW、AOCC、IDDF、CGC等消化领域大会壁报交流。荣获多项Academic Grant、Travel Grant奖励。
版权说明:本文来源于中国医学论坛报“罕见病临床思辨”专栏,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017