


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




概述
N-乙酰谷氨 酸合成酶缺乏症 (N-acetylglutamate synthase deficiency,NAGSD)是一种尿素循环障碍的遗传代谢病,为常染色体隐性遗传。NAGSD患者由于血氨清除障碍导致高氨血症及相关临床表现及并发症。
病因和流行病学
NAGS 基因位于 17q21.31,编码翻译形成 N-乙酰谷氨酸合成酶,该基因突变导致 N-乙酰谷氨酸合成酶缺乏。尿素循环是在肝细胞中发生的一系列化学反应,是将机体各种代谢途径中产生的氨合成尿素并经尿液排出的主要代谢途径(图79-1)。在 N-乙酰谷氨酸合成酶的作用下,乙酰辅酶 A 和谷氨酸转变为N-乙酰谷氨酸和辅酶 A。N-乙酰谷氨酸激活另一种酶-氨甲酰磷酸合成酶,该酶是尿素循环过程中的限速酶(作为尿素循环的第一步,氨和CO2 在该酶的催化下生成氨甲酰磷酸)。因此,N-乙酰谷氨酸合成酶缺乏症患者的N-乙酰谷氨酸不足或者缺乏,导致氨的代谢受阻,血氨升高,引起疾病。
NAGSD 是一种非常罕见的疾病。在世界范围内仅有少数病例报道,总体发病率尚不清楚。
临床表现
与其他尿素循环代谢异常疾病相同,NAGSD 发病的年龄不定,但该病发病年龄往往较早,最常发病的年龄为新生儿时期。大多患儿生后数日内发病,表现为拒奶、呕吐、嗜睡、惊厥、昏迷。部分晚发型患儿以发作性呕吐、嗜睡,伴精神发育异常为特点。高氨血症昏迷的患者可能有脑部伤害与发展迟缓、学习能力受损或智力障碍以及生长发育落后的后遗症。高氨血症患者可能造成颅内压增高、呼吸急促,病情进展会出现呼吸暂停或衰竭,肝肿大。神经系统并发症包括智力障碍、急性高血氨昏迷,甚至死亡。生化改变可见高氨血症、血谷氨酰胺和丙氨酸增高,尿中乳清酸降低或缺如。
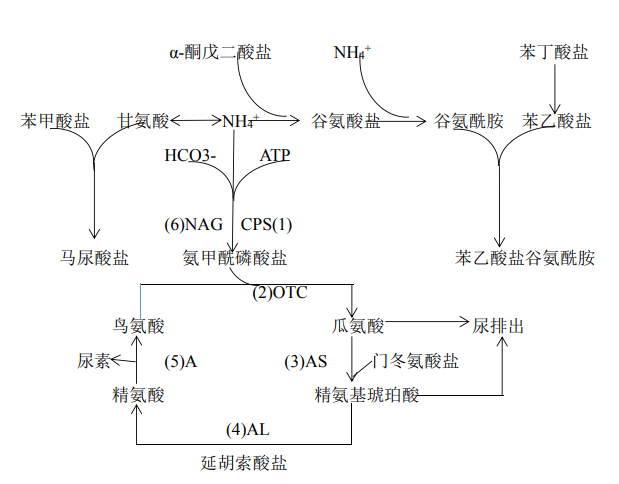
图 79-1 尿素循环
辅助检查
1.一般实验室检查 患者血氨明显增高,可伴有肝酶增高,血糖正常,肌酐和尿素氮降低;血气分析可见呼吸性碱中毒。
2.血氨基酸和尿有机酸分析 血瓜氨酸减低,尿乳清酸阴性。
3.NAGS基因分析 检出 2 个等位基因致病突变可以确诊。
诊断
N-乙酰谷氨酸合成酶缺乏症的诊断主要依据临床症状、生化检查、血氨基酸、尿有机酸和基因检测等结果进行综合分析确定。血氨测定是早期诊断的关键。
临床上对于有神经系统症状伴血氨水平升高的患者,尤其在血气分析示呼吸性碱中毒时,首先考虑尿素循环障碍疾病,进一步行血氨基酸和尿有机酸分析,检出血瓜氨酸水平减低和尿乳清酸减低时,高度提示N-乙酰谷氨酸合成酶缺乏症的可能,确诊有赖于 NAGS 基因分析。
鉴别诊断
1.其他尿素循环障碍性疾病 NAGSD需与其他尿素循环障碍性疾病进行鉴别,如图 79-1 所示,参与尿素循环的 6 种酶依次为 N-乙酰谷氨酸合成酶(NAG)、氨甲酰磷酸合成酶Ⅰ(CPS)、鸟氨酸氨甲酰转移酶(OTC)、精氨酸代琥珀酸合成酶(AS)、精氨基琥珀酸裂解酶(AL)、精氨酸酶(A)。任何一种酶完全性缺乏或部分缺陷,均可造成尿素循环障碍,血氨增高。其中以鸟氨酸氨基甲酰转移酶与氨基甲酰磷酸合成酶Ⅰ的缺乏最常见,临床症状也最重。根据酶缺陷的种类及程度不同,临床表现也有很大不同,但都缺乏特异性。在任何年龄的患者,出现反复呕吐、烦躁、嗜睡、神经行为发育落后和抽搐等,伴血氨明显升高时,均需要考虑该类疾病可能性,需要进一步完善血氨基酸分析。如果精氨酸升高明显,考虑精氨酸酶缺乏症。如果瓜氨酸显著增高,无精氨琥珀酸,考虑精氨琥珀酸合成酶缺乏症。如果瓜氨酸中度增高,有精氨琥珀酸,考虑精氨基琥珀酸裂解酶缺乏症。如果瓜氨酸减少,需要进一步测定尿乳清酸,明显升高提示鸟氨酸氨甲酰转移酶缺乏症,降低应考虑氨甲酰磷酸合成酶缺乏症或N-乙酰谷氨酸合成酶缺乏症,此两种疾病临床表现完全一样,需要通过基因检测而鉴别。
2.有机酸血症 对于高氨血症伴有代谢性酸中毒的患儿,需要考虑有机酸血症。常见的包括甲基丙二酸血症及丙酸血症。鉴别主要是通过尿有机酸分析、血氨基酸和酯酰肉碱谱分析,如尿中检出大量的甲基丙二酸或3-羟基丙酸和甲基枸橼酸,高度提示甲基丙二酸血症或丙酸血症。确诊有赖于基因突变分析。
治疗
1.治疗原则
通过降低蛋白质的摄入,利用氮排出的替代路径纠正高氨血症,降低血氨,同时还要保证正处于生长发育阶段的患儿的营养需求。急性期应立即限制或禁止蛋白质摄入,加强非蛋白类的热量来源。长期治疗应予足够热量、低蛋白饮食。新生儿期发病的、饮食及药物治疗效果不佳者可考虑肝移植。
2.急性期治疗
(1)对症处理:
惊厥者镇静、止惊;脑水肿者甘露醇降颅压;维持水、电解质平衡;积极抗感染;必要时呼吸支持治疗。
(2)限制蛋白质摄入:
以葡萄糖及脂肪为主补充足够热量。静脉葡萄糖用量婴儿期 8~10 mg/(kg·min),儿童可给 10%葡萄糖400~600 ml/m2,脂肪提供热量>80 kcal/(kg·d)。
(3)降血氨治疗:
应用苯甲酸盐、精氨酸、瓜氨酸等,以促进尿素循环,降低血氨水平。根据酶的缺陷不同,应用药物的剂量也不同,当血氨水平持续升高,神经系统损害持续存在时,考虑血液透析或腹膜透析。对于N-乙酰谷氨酸合成酶缺乏症患儿。文献报道口服氨甲酰谷氨酸(N-carbamyl glutamate,NCG)可有效降低血氨,控制病情,但目前国内尚无该产品。
3.长期治疗
长期的低蛋白饮食,摄入量应根据患儿的年龄和病情综合确定,并根据血氨的浓度来调整。小于 6 个月的婴儿 1.5g/(kg·d),学龄前期可1.2~1.5g/(kg·d),学龄期 1g/(kg·d),但是仍有个体差异,需根据生长发育情况及血氨水平调整。
4.其他治疗
部分患儿可考虑肝脏移植治疗。此病的基因治疗尚在研究中。5.遗传咨询 N-乙酰谷氨酸合成酶缺乏症是常染色体隐性遗传病,患者父母再次生育再发风险为 25%。应对所有患者及其家庭成员提供必要的遗传咨询,对高风险胎儿进行产前诊断。
诊疗流程(图79-2)
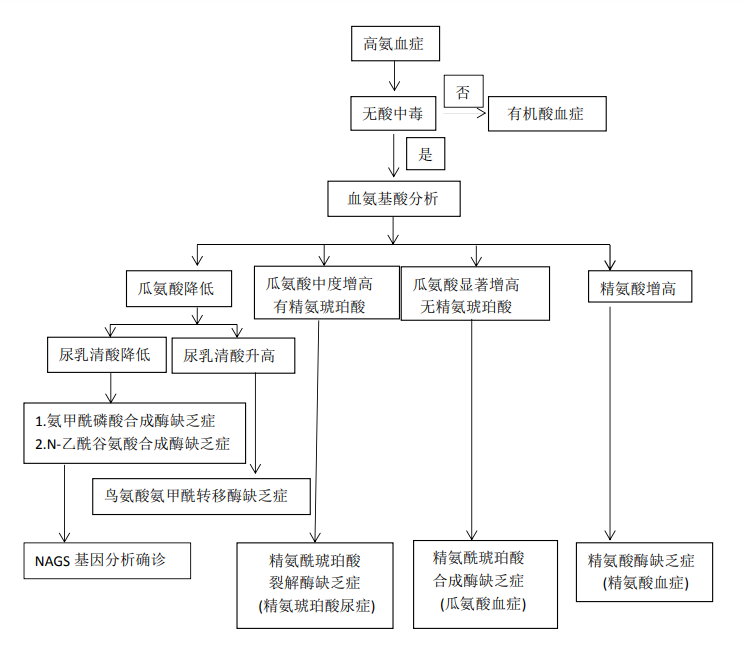
图 79-2 N-乙酰谷氨酸合成酶缺乏症诊疗流程
参考文献(略)
来源:国家卫生健康委员会《罕见病诊疗指南(2019年版)》
罕见病诊疗指南——黏多糖贮积症【心肾系统罕见病】
罕见病诊疗指南——甲基丙二酸血症【神经系统罕见病】
罕见病诊疗指南——羟化酶缺乏症【心肾系统罕见病】
罕见病诊疗指南——Gitelman综合征
罕见病诊疗指南——抗 LGI1 抗体相关脑炎【神经系统罕见病】
罕见病诊疗指南——遗传性痉挛性截瘫【神经系统罕见病】
罕见病诊疗指南——视神经脊髓炎【神经系统罕见病】
罕见病诊疗指南——淋巴管肌瘤病【罕见肿瘤】
罕见病诊疗指南——自身免疫性胰岛素受体病【免疫系统罕见病】
罕见病诊疗指南——原发性肉碱缺乏症【儿童罕见病】
罕见病诊疗指南——原发性肉碱缺乏症【儿童罕见病】
罕见病诊疗指南——β-酮硫解酶缺乏症【儿童罕见病】
罕见病诊疗指南——生物素酶缺乏症【神经系统罕见病】
罕见病诊疗指南——自身免疫性垂体炎【神经系统罕见病】
罕见病诊疗指南——心脏离子通道病【心肾系统罕见病】
罕见病诊疗指南——天使综合征【神经系统罕见病】
罕见病诊疗指南——自身免疫性脑炎【神经系统罕见病】
罕见病诊疗指南——抗NMDAR脑炎【神经系统罕见病】
罕见病诊疗指南——白化病【呼吸和皮肤系统罕见病】
罕见病诊疗指南——精氨酸酶缺乏症【神经系统罕见病】
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017