




200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




新基因、新表型
来自埃森大学医学院的Andreas Roos作了题为“Bi-alleic variants of FILIP1 cause congenial myopathy, dysmorphism and neurological defect”的报告,展示了细丝蛋白-A相互作用蛋白1(FILIP1)基因突变的临床表型及致病机制。
在4个家系的5个肌病患者中发现存在FILIP1基因纯合变异(2个错义突变和2个无义突变)。携带纯合p. Pro1133Leu突变的患者临床表现为脑部畸形、神经发育迟缓和肌肉无力;肌肉活检超微结构可见管样肌原纤维排列紊乱、自噬空泡形成和FlnC异常聚集。进一步的蛋白组学研究证明了其致病性:位于神经元和肌细胞内的结构蛋白FILIP1发生p. Pro1133Leu突变后不仅自身稳定性下降,还可导致FLNc在内的多种与FILIP1互相作用的蛋白表达异常,最后引起广泛神经系统受累。
来自美国国立卫生研究院的Rotem Or Bach作了题为“Impaired iron-sulfur cluster assembly due to biallelic variants in CIAO1 leads to a novel muscle disease”的报告,展示了CIA关键组成部分CIAO1基因突变在神经肌病中的致病作用。
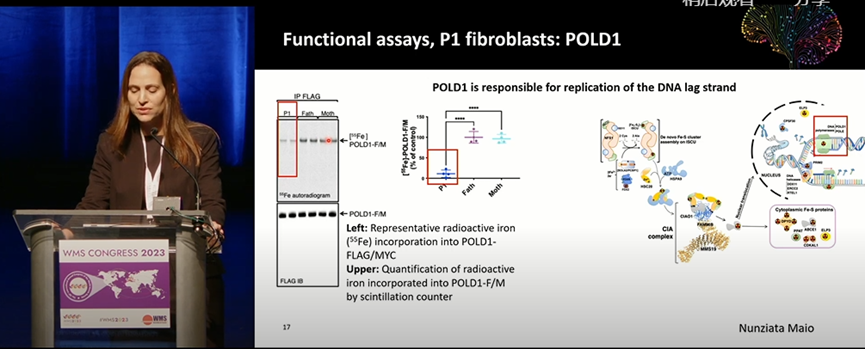
在4例不相关的肌病患者中发现CIAO1基因复合杂合突变(P1:p.H302P/p.F250_L339del;P2 和P3:p.H302P/p.R65W;P4:p.D171G/p.H251L)。4例患者均表现为青少年早期起病的进行性中轴肌和近端肌无力、肌酸激酶升高和呼吸功能不全。2例患者中还存在学习困难、自闭倾向、大细胞贫血和胃肠道等非肌肉症状。肌肉MRI提示弥漫性轻度肌萎缩和脂肪浸润,近端肌受累明显。肌肉活检显示肌病和轻度肌营养不良样改变;氧化染色可见明显的线粒体聚集。P1肌组织在电镜下可见巨大且形态异常的线粒体;肌肉蛋白印迹和生化检测均表明其线粒体呼吸链功能障碍。P1来源的成纤维细胞中也证实存在与核基因组的复制和维持相关的铁硫蛋白合成障碍和线粒体功能受损;慢病毒介导的CIAO1表达可挽救细胞表型。
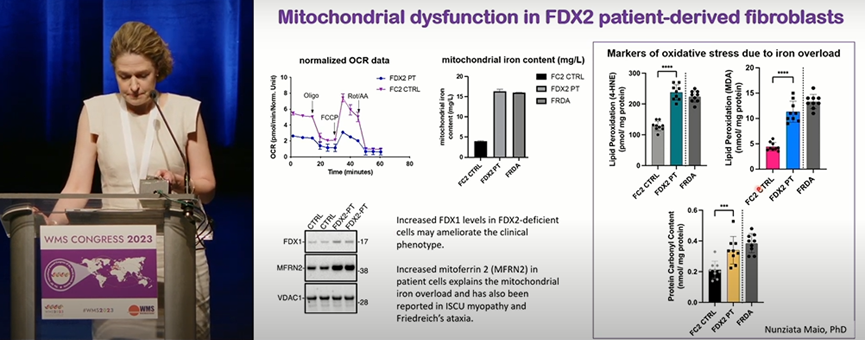
来自美国国家卫生研究院的Reghan Foley带来了CIA途径的另一致病基因ferredoxin 2(FDX2)突变所致的病例。该患者携带FDX2杂合突变(c.271C>T, p.L91F;c.344A>G, p.H115R)。患者17个月时发生失明,临床和肌肉影像学上均存在肌病特征。通过患者来源成纤维细胞的体外研究发现FDX2蛋白突变后,其与CIA成分之间的相互作用发生异常,导致线粒体氧化磷酸化功能异常。在治疗方面,患者3岁时开始服用艾地苯醌,5岁时改用辅酶Q10类似物(MitoQ)。经治疗后,患者的肌力和行走能力呈平稳改善,但视力始终未恢复。
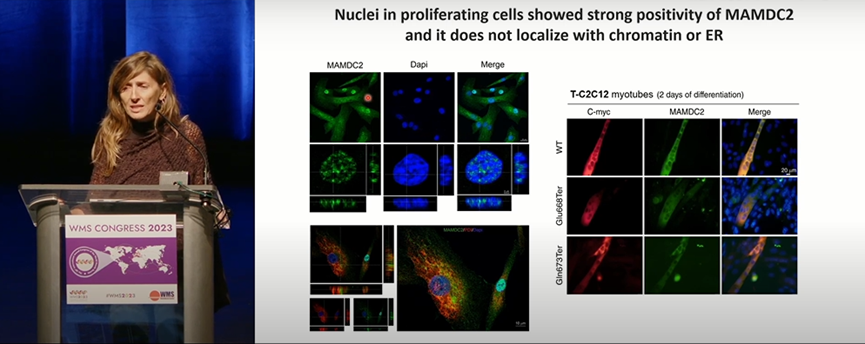
来自塞维利亚生物医学研究所的Paradas C分享了MAMDC2基因在肌营养不良中的致病作用。
作为潜在的细胞外基质(ECM)蛋白,MAMDC2在肌肉发育和ECM中的作用尚未明确。Paradas C等在来自两个家系的17名常染色体显性肌营养不良患者中检测到位于MAMDC2基因最后一个外显子的杂合截短突变,并在家系中共分离。肌肉MRI提示肌肉受累特征类似COL6突变相关肌病。Paradas C团队证实MAMDC2蛋白在成人骨骼肌中表达,且定位于胞浆和肌核,并被分泌到ECM。由于最后一个外显子编码的无序区域具有极性残基的组成偏差,该截短突变可能会导致突变蛋白具有毒性效应。
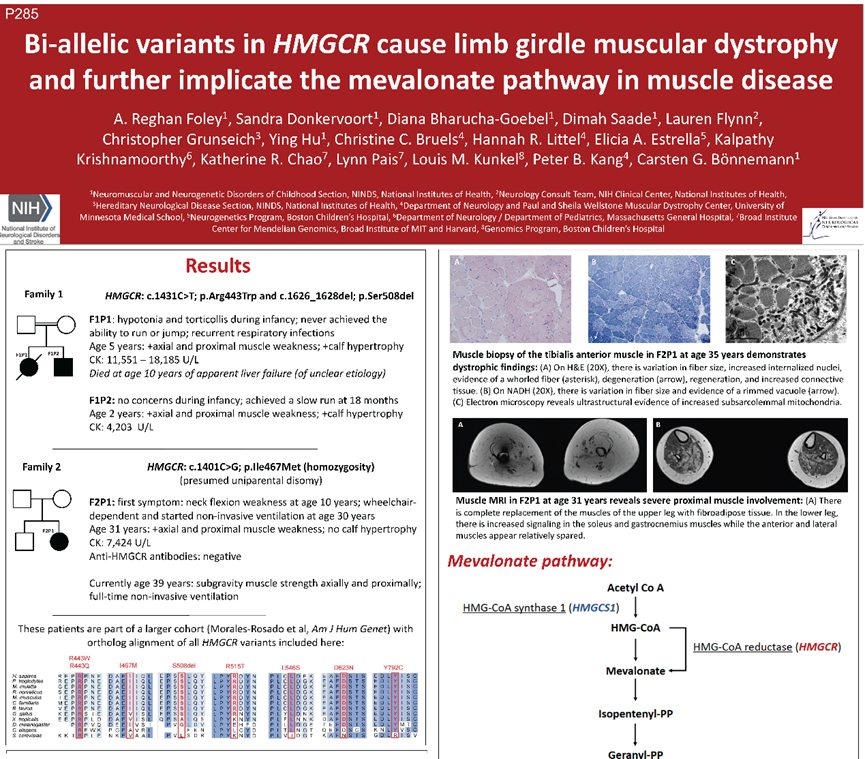
甲羟戊酸途径(Mevalonate pathway)是以乙酰辅酶A为原料合成异戊二烯焦磷酸和二甲烯丙基焦磷酸的一条代谢途径,其中HMGCR编码 3-羟基-3-甲基戊二酰辅酶A还原酶,后者作为该途径的限速酶,是不少他汀类药物的分子靶点。此前,HMGCR纯合变异已被证实为甲羟戊酸内酯治疗有效的LGMD的病因。来自美国国立卫生研究院的A. Reghan Foley等报道了来自2个不相关家系的3例HMGCR基因变异患者(F1:c.1431C>T/c.1626_1628del;F2:c.1401C>G纯合变异),为新生儿或儿童起病,表现为进行性近端肌无力和呼吸功能不全,CK值显著升高,肌肉MRI和肌活检结果均显示肌营养不良样改变。该研究扩大了HMGCR基因突变临床表型谱,并进一步提示甲羟戊酸途径在肌肉疾病发表中的重要性。
荷兰拉德堡德大学医学中心团队对NEM6开展了一项单中心横断面研究,旨在帮助临床医生识别NEM6以及加强该类患者的临床护理。该研究采用多项综合性评估,包括(1)病史;(2)体格检查;(3)问卷:SF-36(用于评估生活质量)和CIS(用于检查疲劳严重度)量表;(4)功能性测试:Mini-BESTest(用于评估平衡控制)、MFM(用于测量运动功能)、6MWT(用于测量步行距离)等;(5)经颅磁刺激(TMS):测量正常化峰值松弛率(NpRR)来评估肌肉松弛的动力学。根据问卷统计结果,大部分NEM6患者除了全身性肌无力和轻度功能测试异常之外,还存在生活质量下降、疲劳严重程度增加以及跌倒发生率增加等一系列生活困难。
来自比利时的安特卫普大学团队报道了7例SPTAN1(编码Alpha-II-Spectrin蛋白)基因杂合移码突变相关的远端肌病,患者同时合并智力障碍、面瘫和小头畸形等神经源性特征。CT/MRI影像学结果显示近远端肌肉具有分裂征。肌肉活检可见肌纤维大小不等、分裂纤维以及肌间纤维化等肌病改变。功能学实验显示突变的Alpha-II-Spectrin蛋白表达与正常蛋白无明显异常,但其mRNA水平下降40%,该移码突变导致远端无力和智力障碍的具体致病机制有待进一步研究。
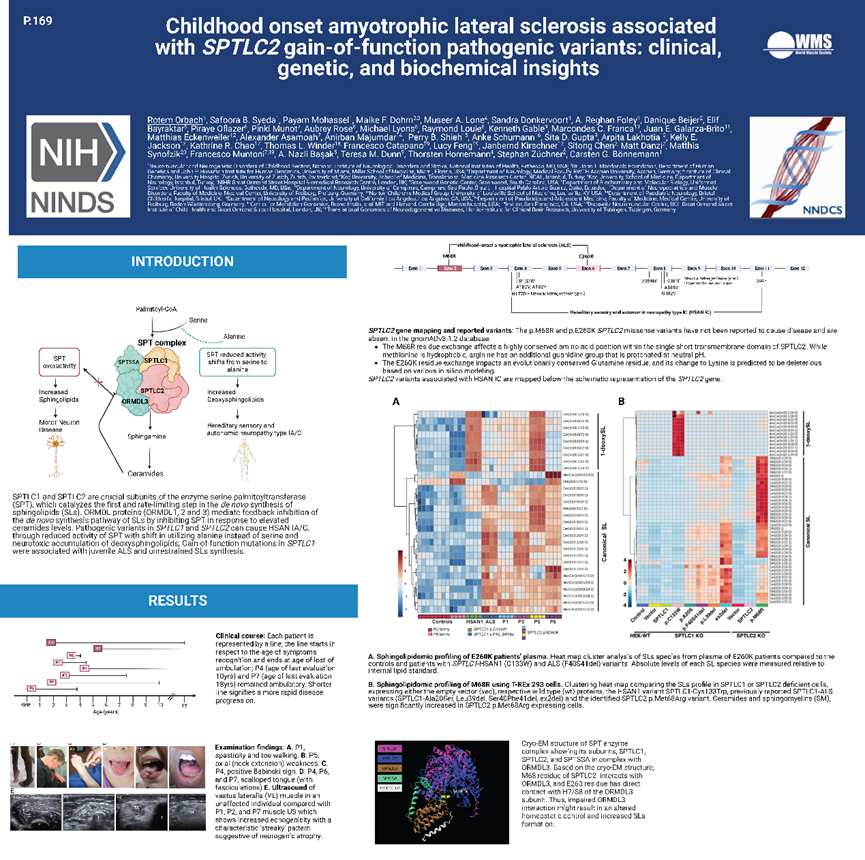
SPTLC1和SPTLC2是丝氨酸棕榈酰转移酶(SPT)的关键亚基,后者作为催化鞘脂类脂质(sphingolipids,SLs)从头合成的第一步,也是限速步骤。SPTLC1和SPTLC2的致病性变异可通过降低SPT的活性,使得丙氨酸代替丝氨酸,以及脱氧鞘脂的神经毒性累积,导致Ⅰ型遗传性感觉和自主神经病(HSAN IA/C)。美国国立卫生研究院报道了8例携带SPTLC2基因新突变的ALS病例。发病年龄为新生儿至青少年,临床上同时累及上、下运动神经元,而无感觉神经元受累,5/7患者因呼吸肌无力需要呼吸支持。基因检测提示病例1-6携带c.778G>A (p.E260K)突变,病例7、8携带c.203T>G (p.M68R)突变。根据SPT酶复合物的冷冻电镜结构,预测p.E260K和p.M68R突变都会影响SPTLC2与抑制亚基ORMDL3的相互作用。鞘脂组学分析显示p.E260K患者来源血浆和p.M68R突变的HEK 293细胞中的SLs水平显著升高。该研究证实了SPTLC2与儿童起病ALS的相关性,并进一步确定了鞘磷脂代谢异常可参与运动神经元病发病。
新生物标志物
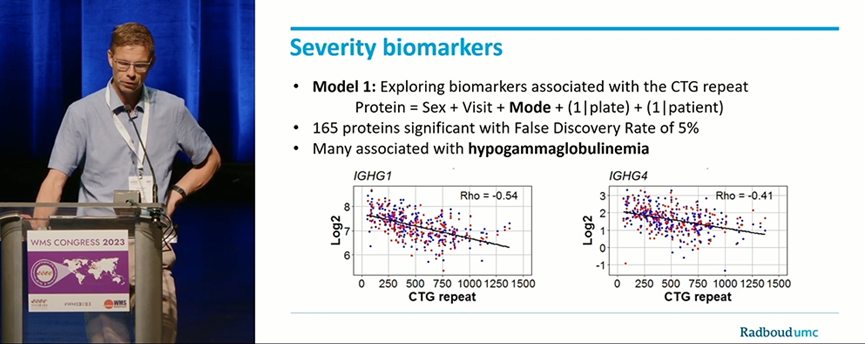
拉德堡德大学医学院P.'t Hoen对针对DM1的多中心随机对照试验(OPTIMISTIC trial)中450例成年DM1患者的外周血进行基于质谱的蛋白质组分析。至少80%的标本中可检测到来自263个蛋白组的2702种独特多肽。通过线性混合效应模型和基于机器学习的算法,鉴定出与CTG重复次数和运动功能(六分钟步行测试,6MWT)相关蛋白。该研究中共鉴定出167组蛋白与CTG重复次数相关,73组蛋白与运动功能相关,其中47组蛋白与两者均相关。值得注意的是,免疫球蛋白水平下降与CTG重复次数相关;运动功能减退与补体水平升高相关。上述成果为未来临床试验和治疗策略提供了潜在的生物标志物。
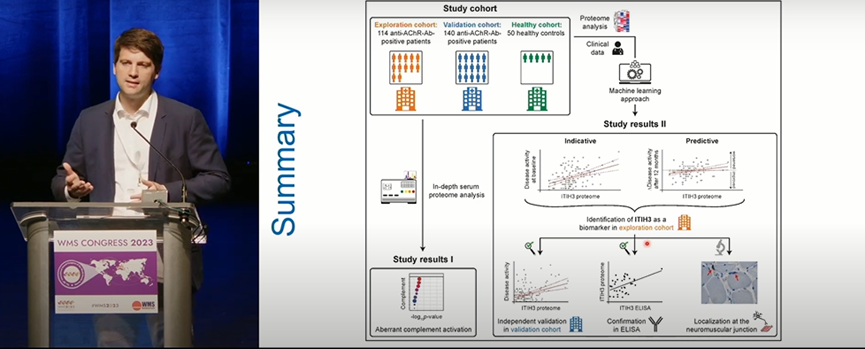
目前,MG的治疗主要取决于临床特征的评估,这可能受到症状波动和医生主观判断的影响。因此,发现新的生物标志物以客观评估和预测MG的病情仍然是一个迫切的需求。海因里希·海涅大学团队通过对AChR-Ab阳性MG患者血清进行蛋白质组学分析合机器学习算法,发现ITIH3可作为MG的潜在生物标志物,并运用ELISA技术在140例MG病例的前瞻性队列中获得了验证。相比正常对照,MG患者的神经肌肉接头中ITIH3显著高表达,为今后MG血清学新发现打下了病理生理学基础。
撰稿:章佳隆 复旦大学附属华山医院神经内科
审核:奚剑英、朱雯华 复旦大学附属华山医院神经内科
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017