


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:东部战区总医院 国家肾脏疾病临床医学研究中心 郭锦洲 赵亮 黄湘华 刘志红
病史摘要
主诉 患者男性,71岁,因“多浆膜腔积液3年余,发现红细胞增多及肾周积液1月余”于2021年2月1日入院。
现病史 2017年底患者体检查胸腹部CT示右侧胸腔积液,腹膜后多处渗出,血白蛋白、肌酐正常,自身抗体阴性,正电子发射计算机断层显像(PET-CT)未见明显肿瘤病灶,未特殊处理。2019年1月复查PET-CT示双侧胸腔、腹腔和盆腔积液,全身未见明确恶性高代谢病灶,血常规示血红蛋白220g/L,红细胞压积64.5%,血免疫固定电泳阳性(未见报告单),骨髓细胞学未见明显异常,胸水脱落细胞学未见恶性细胞,胸水流式细胞学检查示淋巴细胞占88%,CD4/CD8比例大于9∶1,余抗原表达未见异常,血管超声示左侧颈内静脉及左锁骨下静脉近端血栓形成,考虑真性红细胞增多症(PV),予华法林抗凝及羟基脲0.5g/d治疗,门诊根据血红蛋白水平调整羟基脲剂量。2019年5月查人红细胞生成素(EPO)74.5mIU/ml,未特殊处理。2020年12月24日查肾脏超声示双侧肾周局限性大量积液,尿蛋白±,血常规示血红蛋白146g/L,白细胞、血小板正常,血生化示白蛋白32.7g/L,肌酐93.3μmol/L,体液免疫示IgG30.5g/L,补体C30.754g/L,C4正常,骨髓穿刺细胞学示粒系、红系、巨核系增生减低,血小板散在可见;骨髓活检示骨髓增生减低(20%),粒红比大致正常,粒系以中性中幼粒细胞及以下阶段为主,红系增生减低,以中晚幼红为主。全腹部CT平扫示双肾周低密度影,右侧输尿管上段扩张。心电图:窦性心律,肢导联低电压。2021年1月7日行双肾包膜下积液穿刺引流术,引流液生化示尿素8mmol/L,肌酐105μmol/L(同时查随机尿生化示尿素173mmol/L,肌酐11126μmol/L),24h引流液蛋白定量10720mg(总量800ml),引流液肿瘤标志物、细菌培养、结核杆菌DNA、脱落细胞学检查未见异常,引流液流式细胞术检查示淋巴细胞占93.8%,抗原表达未见异常。腹壁皮肤脂肪活检示刚果红染色阳性,偏振光镜下见苹果绿样折光物沉查,考虑系统性淀粉样变性,建议化疗。患者至我科门诊就诊后,于2021年2月1日收住入院。患者约3年前出现前胸部皮肤毛细血管扩张,未予重视。患者起病以来精神、食欲、睡眠尚可,大便正常,尿量正常,体重下降10kg左右,体力有所下降。
既往史 患者既往20多岁时因“急性阑尾炎”行阑尾切除术。吸烟50年,每天约40支,饮酒50年,白酒约200ml/d,已戒烟、戒酒2年。婚育史、家族史无特殊。
体格检查
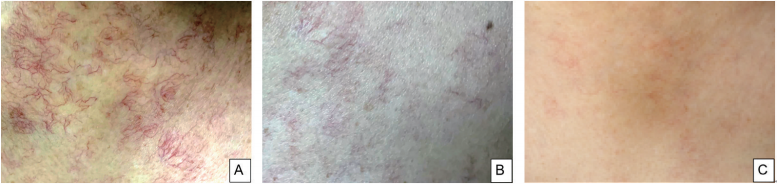
体温37℃,脉搏64次/min,呼吸16次/min,脉氧98%,血压137/82mmHg,颈部、双侧肩部、胸前区皮肤可见毛细血管扩张(图1A),心肺未见异常,腹软,右下腹陈旧性手术疤痕,全腹无压痛、反跳痛,无肾区叩击痛,移动性浊音阴性,双下肢无浮肿。
图1 胸前区皮肤毛细血管扩张化疗前后的变化
A:胸前区皮肤毛细血管扩张;B:化疗4个疗程后胸前区皮肤毛细血管扩张明显减少;C:化疗7个疗程后胸前区皮肤毛细血管扩张基本消失。
实验室检查
尿液 尿常规:蛋白阴性(-)、红细胞阴性(-)。尿白蛋白/肌酐比值53.5mg/g。尿N-乙酰-β-D-氨基葡萄糖苷酶(NAG)3.4U/g·cr,视黄醇结合蛋白(RBP)0.12mg/L。
血液
血常规:白细胞计数4.04×109/L,血红蛋白136g/L,血小板计数232×109/L,C反应蛋白6.5mg/L。
血生化:尿素20.91mg/dl,肌酐1.01mg/dl,尿酸311μmol/L,估算肾小球滤过率(eGFR)75ml/min,白蛋白34.10g/L,球蛋白34.6g/L,电解质、转氨酶、血脂正常。
κ血游离轻链(FLC)22.8mg/L(参考值3.3~19.4mg/L),λFLC30.8mg/L(参考值5.71~26.3mg/L),κ/λ0.74(参考值0.26~1.65)。
血免疫球蛋白亚类IgG41150.0mg/L。免疫固定电泳见κ型IgG单克隆免疫球蛋白条带。
血管内皮生长因子(VEGF)302.15pg/ml(参考值0~142.2)。
外周血全外显子基因检测:未发现临床表型高度相关同时致病性证据较为充分的基因变异。空腹血糖、甲状腺功能正常。
辅助检查
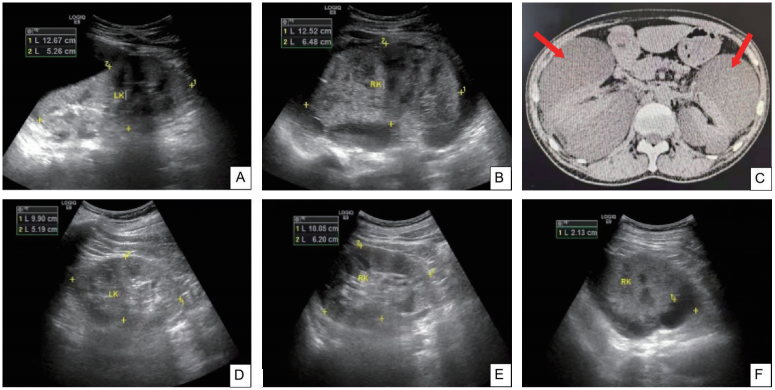
肾脏超声 左:126mm×55mm×63mm,右:125mm×64mm×62mm。双肾体积增大,结构不清楚(图2A、图2B);双肾周可探及大范围无回声区。
右心声学造影 经左肘静脉注入造影剂后,右房、右室依次显影,16个心动周期后左房、左室可见少许造影剂气泡回声,不随胸腹腔压力变化而改变。诊断为小肺动静脉分流。
胸腹部CT 两肺小结节,考虑良性结节可能;右肺上、下叶少许纤维条索;左侧胸腔积液;双肾肾周大量积液(图2C)。
骨髓 形态:大致正常骨髓象。流式细胞术(浆细胞):异常克隆性浆细胞占有核细胞的0.3%,表型:CD38强表达,cκ、CD117、CD138、CD81表达,CD45弱表达,cλ、CD19、CD56、CD27不表达;另见0.28%正常多克隆性浆细胞。流式(淋巴细胞):未见异常淋巴细胞。荧光原位杂交:未见检测位点遗传学异常。
病理 腹壁皮肤脂肪组织刚果红染色阴性。
图2 双肾及肾周积液化疗前后的影像学变化
A、B:肾脏超声示双肾体积增大,结构不清楚;C:双侧肾周大量积液(→);D:化疗7个疗程后左肾肿大缓解,左侧肾周积液消失;E:化疗7个疗程后右肾肿大缓解;F:化疗7个疗程后右侧肾周积液明显减少(最大深度21mm)。
诊断与治疗
患者诊断为TEMPI综合征。
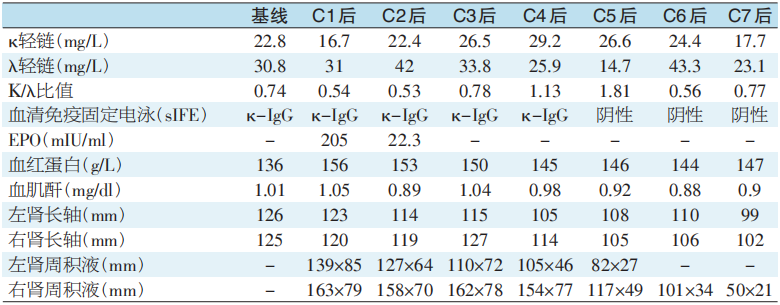
患者于2021年2月19日、3月20日、5月8日、6月11日、7月20日、8月24日、10月13日、11月20日行8个疗程BD方案化疗(硼替佐米1.3mg/m2+地塞米松20mg,d1、4、8、11),2个疗程后复查EPO正常(表1),5个疗程后复查血清免疫固定电泳阴性,皮肤毛细血管扩张逐渐减轻(图1B、图1C),左侧肾周积液消失(图2D),右侧肾周积液明显减少(图2E、图2F)。
表1 患者治疗前后血液检验和肾脏大小、肾周积液的变化(C1为第1疗程,C2~C7以此类推)
诊疗分析
71岁男性患者,病程3年余,病史特点如下:①体检发现胸、腹腔积液,多次查PET-CT、肿瘤标志物、胸水相关检查均未找到明确肿瘤证据;②病程中发现红细胞增多伴EPO升高,左侧颈内静脉及左锁骨下静脉近端血栓形成,外院诊断为“真性红细胞增多症”,给予羟基脲和抗凝治疗,红细胞降至正常范围;③近期出现双侧肾周大量积液;④查体可见颈部、双侧肩部、胸前区皮肤毛细血管扩张。
该患者外院诊断为PV,羟基脲治疗有效。PV是一种造血干细胞的克隆性慢性骨髓增殖性疾病,进展缓慢,以血栓和出血为主要临床表现,大约95%的PV患者存在JAK2基因突变。诊断PV必须有JAK2突变或者血清EPO浓度低于正常参考值水平,同时排除继发因素。本例患者基因检测未见JAK2基因突变,且EPO升高,可排除PV,考虑EPO升高导致红细胞增多。EPO是一种人体内源性糖蛋白激素,主要由肾脏产生,少量自肝脏产生,缺氧可刺激EPO产生。本例患者无心肺功能不全,脉氧正常,临床上无缺氧表现,红细胞增多和EPO升高早于双侧肾周积液,故不考虑缺氧导致EPO升高。文献报告过量注射EPO并未引起多浆膜腔积液,且本例患者合并单克隆免疫球蛋白(M蛋白)血症,故不能单用EPO升高来解释本例患者的临床表现。
M蛋白血症患者具有与M蛋白相关,或者与产生M蛋白的克隆性B细胞或浆细胞相关的临床表现,如肾脏、皮肤、神经或其他器官损害,称为有临床意义的单克隆免疫球蛋白血症(MGCS)。本例患者尿检基本正常,eGFR符合老年男性特点,不考虑肾脏受累;无中枢及周围神经受累表现,不考虑神经受累;外院皮肤组织刚果红染色阳性,我院行重复皮肤活检排除皮肤淀粉样变性,但查体可见多处皮肤毛细血管扩张,考虑有皮肤受累的MGCS。累及皮肤的MGCS包括冷球蛋白血症、施尼茨勒综合征、毛细血管渗漏综合征、硬化性黏液水肿、渐进性坏死性黄肉芽肿、POEMS综合征、TEMPI综合征。
冷球蛋白血症常继发于感染性疾病、自身免疫性疾病和免疫增生性疾病。冷球蛋白遇冷在微细血管内沉积,引起远端缺血产生临床症状,此外冷球蛋白作为免疫复合物可激活补体,引起血管炎。除原发疾病的临床表现外,其他常见症状包括雷诺征、皮肤紫癜、坏死、溃疡、寒冷性荨麻疹、关节痛、感觉麻木等,以及深部血管受累时所涉及的肾、脑、肝和脾等器官损害。本例患者不符合冷球蛋白血症的典型皮肤损害。
施尼茨勒综合征是一类获得性自身炎症性疾病,多见于成年男性,临床表现为慢性荨麻疹、发热、骨痛,有时可触及淋巴结、脾或肝肿大,血检可见血沉增快、C反应蛋白升高和单克隆IgM-κ。疾病发生可能与NLRP3基因突变造成IL-1β大量生成有关,靶向IL-1单抗治疗有效,M蛋白在疾病中的作用机制不明。本例患者临床表现不符合施尼茨勒综合征。
毛细血管渗漏综合征,于1960年由克拉克森医生首次报告,低血压、血液浓缩和低蛋白血症是诊断的关键。68%的成年患者存在M蛋白,常见的是IgG-κ。本例患者无周期性低血压休克,无皮肤水肿,不符合。
硬化性黏液水肿常表现为广泛性丘疹和硬皮病样皮疹,常伴有单克隆IgG,除皮肤外还可累及神经、关节、胃肠道和心脏。皮肤病理可见真皮层黏蛋白沉积、胶原蛋白增厚和成纤维细胞增生。本例患者皮肤损害不符合硬化性黏液水肿。
渐进性坏死性黄肉芽肿是一种非朗格汉斯细胞组织细胞增多症,好发于老年人,通常与浆细胞疾病或淋巴增生性疾病分泌的M蛋白有关。典型皮损为眼睑出现黄色至橙色的丘疹、斑块和/或结节,也可见于面部、躯干和四肢。皮肤病理见真皮和皮下组织内毛玻璃样渐进性坏死,外围有组织细胞、泡沫细胞、托顿(Touton)巨细胞、异物巨细胞和其他炎细胞浸润。本例患者皮肤损害不符合渐进性坏死性黄肉芽肿。
POEMS综合征是一种与浆细胞病有关的多系统病变,临床上以多发性周围神经病、脏器肿大、内分泌障碍、M蛋白血症和皮肤病变为特征。50%~90%的患者有皮肤改变,其中以局灶性或全身性皮肤色素沉着最常见,其他表现有水肿、多毛、多汗、杵状指、雷诺现象、血管瘤及白甲等。本例患者无多发性周围神经病、脏器肿大,无糖尿病、甲减、阳痿等内分泌障碍,皮肤改变也不符合POEMS综合征。
TEMPI综合征是一类罕见的浆细胞疾病,2011年首次被学者提出。临床表现为以下五联征:毛细血管扩张(T),EPO浓度升高和红细胞增多(E),单克隆免疫球蛋白血症(M),肾周积液(P),肺内分流(I)。进一步完善右心声学造影证实存在小肺动静脉分流。本例患者符合TEMPI综合征的五联征。
讨论
//
TEMPI综合征的表现和流行病学
1994年英国学者伯顿等报告1例高加索男性患者,1978年(35岁)体检发现高血压、红细胞增多伴EPO升高、血肌酐升高(140μmol/L),12年后发现双侧肾周积液,穿刺液生化分析提示淋巴液,作者认为可能是双侧淋巴管扩张继发红细胞增多。此后也有类似临床表现的零星个案报告。2010年来自麻省总医院的医疗团队报告了1例中年男性患者,自2003年起先后发现红细胞增多、EPO升高、血M蛋白阳性(κ-IgG)、皮肤和口咽部毛细血管扩张、右侧颈内静脉血栓、双侧胸腔积液、双侧肾周积液。该患者考虑为肾淋巴管扩张继发红细胞增多,同时合并意义未明的单克隆免疫球蛋白血症(MGUS)。该文章发表后作者收到同行分享的类似病例,并结合文献检索,于2011年提出TEMPI综合征的概念。
TEMPI综合征的典型临床表现包括毛细血管扩张、红细胞增多伴EPO升高、M蛋白血症、肾周积液和肺内分流,此外还可能出现静脉血栓、肝血管瘤、自发颅内出血等。截至目前文献报告仅有20多例TEMPI综合征患者,多于40~60岁发病,无性别、种族或地区差异。绝大多数患者首发表现为红细胞增多和毛细血管扩张,部分患者被误诊为PV而接受静脉切开放血治疗。EPO水平升高,无JAK2基因突变,以此与PV鉴别。毛细血管扩张主要见于面部、上背部、胸部和手部,下肢少见。毛细血管扩张不限于体表皮肤,有文献报告1例患者行结肠镜检查示多发小血管扩张。M蛋白血症是TEMPI的特征之一,以κ型IgG多见,而POEMS综合征的M蛋白以λ型 IgG或IgA为主。TEMPI综合征虽有M蛋白血症,但血游离轻链数值及比值正常或接近正常,骨髓中浆细胞比例一般不超过10%。随着疾病进展,EPO水平逐渐升高,部分患者超过检测上限(>5000mIU/ml),进一步加重红细胞增多症。肺内动静脉分流在高分辨率CT扫描无法显影,此时可采用右心声学造影,右心显影后数个心动周期后在左心观察到微泡(本例患者采用该方法明确存在肺内动静脉分流)。99Tcm-大颗粒聚合白蛋白(99Tcm-MAA)核素显像能更好地显示肺内分流。肺内分流可导致静息状态血氧饱和度下降,随着疾病进展最终需要吸氧治疗。肾周积液的病因尚不清楚。有学者发现肾周积液穿刺液理化性质均与血浆接近,但蛋白质含量仅1g/L,肾活检病理示肾小球、肾小管和管周毛细血管基本正常,小动脉周围淋巴管扩张,作者认为淋巴管扩张导致肾周积液,肾脏受压继发EPO升高。本例患者符合TEMPI综合征的五项临床表现,肾周积液的蛋白质浓度(13.4g/L)显著高于文献报告,约占血液中蛋白质含量的20%,支持淋巴液来源。
//
TEMPI综合征的发病机制
TEMPI综合征的发病机制目前尚未明确。起初人们认为是由于肾脏淋巴管扩张导致肾周积液,肾脏受压后缺血缺氧从而引起EPO升高,但该理论不能解释骨髓中单克隆浆细胞和M 蛋白血症,且部分患者EPO升高早于肾周积液。在临床缓解后再次复发的患者,最早观察到的是EPO升高,紧随其后出现M蛋白和皮肤毛细血管扩张。但文献报告过量注射EPO并未引起TEMPI综合征的其他症状,因此EPO不考虑为关键致病因素。
本例患者查VEGF轻度升高,VEGF在POEMS综合征的发病机制中发挥重要作用,VEGF升高是POEMS综合征的主要诊断标准之一,VEGF达到完全缓解(CR)的患者可获得更长的无进展生存期(PFS)和总体生存期(OS);而文献报告TEMPI综合征患者VEGF水平正常,且靶向VEGF的贝伐单抗治疗效果不佳,故不考虑VEGF参与TEMPI综合征的发病机制。
靶向浆细胞治疗取得很好的临床疗效,随着M蛋白转阴,临床症状可获得完全缓解。因此有人提出可能是浆细胞分泌的某种循环因子参与疾病机制,或者其分泌的M蛋白以自身抗体的形式导致自身免疫性疾病。既往研究表明,多发性骨髓瘤(MM)浆细胞分泌巨噬细胞移动抑制因子(MIF),可促进浆细胞向骨髓黏附,调节骨髓瘤细胞的分化和去分化。有学者发现MIF通过诱导VEGF和EPO的表达在视网膜新生血管形成中发挥重要作用。另有学者发现MIF可以上调低氧诱导因子1α的表达水平和转录活性,从而促进EPO产生。
MIF通过上调诱导型一氧化氮(NO)合成酶的表达促进NO的产生,参与肺内分流和毛细血管扩张的形成。此外MIF能促进胸腹腔积液形成,也可能促进肾周积液的产生。还有通过全基因组测序(WGS)发现TEMPI综合征患者的单克隆浆细胞具有22q11.23基因重复,而该位点对应MIF基因。TEMPI综合征患者的骨髓浆细胞MIFmRNA表达和骨髓中MIF水平显著高于健康对照人群,血液中MIF水平高于MM患者和健康对照。随着靶向浆细胞治疗方案的进行,患者血液中的血红蛋白、M蛋白定量、EPO和MIF水平同步下降,此时患者的临床症状包括毛细血管扩张、肾周积液和肺内分流均有缓解,提示MIF参与或主导了TEMPI综合征的发病机制,而22q11.23基因重复和MIF表达上调可能是TEMPI综合征的主要病理生理机制。
//
TEMPI综合征的治疗
早期由于对疾病认知不足,TEMPI患者仅能接受对症治疗,红细胞增多时定期接受静脉切开放血,最终导致医源性缺铁性贫血;肾周积液则接受腹腔引流手术;肺内动静脉分流、严重缺氧患者需接受吸氧治疗。随着TEMPI综合征概念的提出,尤其观察到所有患者均有M蛋白血症,靶向浆细胞的多种治疗方案逐渐尝试用于TEMPI综合征的治疗。硼替佐米化疗和马法兰预处理序贯自体造血干细胞移植的治疗方案均能获得血液学部分缓解(PR)以上,甚至CR的疗效。即使以上方案未取得临床疗效,来那度胺和达雷妥尤单抗同样取得临床疗效。本例患者经BD方案化疗后,获得血M蛋白转阴、临床症状明显好转的临床疗效。
小结
临床遇到难以解释的红细胞增多、肾周积液伴M蛋白血症时,应考虑TEMPI综合征可能,需要注意查体和完善检查明确是否存在皮肤毛细血管扩张和肺内动静脉分流。靶向浆细胞的多种治疗方案均适用于TEMPI综合征的治疗。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017