


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




概述
多种酰基辅酶 A 脱氢酶缺乏症(multiple acyl-CoA dehydrogenase deficiency,MADD),又称为戊二酸血症Ⅱ型(glutaric acidemia Ⅱ)或戊二酸尿症Ⅱ型(glutaric aciduria Ⅱ),是基因缺陷导致线粒体中电子传递黄素蛋白(ETF)或电子传递黄素蛋白脱氢酶(ETFDH)功能障碍所致的脂肪酸氧化代谢紊乱。属于常染色体隐性遗传病。
病因和流行病学
线粒体脂肪酸β氧化是机体重要的能量来源,ETF、ETFDH 是线粒体呼吸链中电子传递的关键转运体。其中,ETF 位于线粒体基质内,接受多种脱氢酶脱氢产生的电子,转运至内膜的 ETFDH,最后转运至复合体Ⅲ,产生ATP。ETF由两个亚基组成,分别为 ETFA 和 ETFB。编码 ETFA、ETFB 和ETFDH基因的突变阻碍了还原当量的流动,氧化磷酸化受阻,导致脂肪酸、支链氨基酸、维生素B 和能量代谢障碍,代谢产物沉积。
目前 MADD 的发病率未见明确报道,亦未见明确的热点突变报道。
临床表现
MADD 的症状谱具有交叉性,主要分 3 种类型:MADDⅠ型为新生儿期起病,伴先天畸形;MADDⅡ型为新生儿期起病,不伴先天畸形;MADDⅢ型为晚发型。
新生儿期起病常致命,可表现为代谢危象,包括代谢性酸中毒、非酮症性低血糖、高氨血症,其他特征包括多系统受累(肌张力低下、心肌病、肝肿大),排泄大量脂肪酸和氨基酸衍生的代谢产物、“汗脚”味体臭、昏迷、新生儿期死亡。快速进展的肥厚性心肌病常见于新生儿期起病的类型,猝死概率非常大,即使经过新生儿筛查在早期即检测出有此病,也难以逆转。
先天畸形被认为是未被胚胎转运纠正的那部分有毒代谢物累积的后果。脂肪酸氧化代谢受阻的程度决定了子宫内的酸中毒程度,进而阻碍正常的胚胎形成。先天畸形包括增大的多囊肾、摇篮底足(rocker-bottom feet)、下腹部肌肉缺失、尿道下裂和阴茎下弯,肌张力低下、脑皮质发育不良和胶质增生。其他畸形特征包括眼距过宽、耳畸形、巨颅、前囟过宽、高前额、鼻梁低平。这些特征和严重的肉碱棕榈酰转移酶Ⅱ缺乏(carnitine palmitoyl-transferase 2 deficiency,CPT2D)相似。
晚发型 MADD 的发病年龄和症状在不同病例中差异很大,主要表现为不伴有先天畸形,但终身伴有急性间歇发作的呕吐、脱水、低酮性低血糖、酸中毒及意识障碍的风险。年龄大者可有肝脏肿大或脂质沉积性肌病。急性失代偿发作可由感染、发热、手术、低能量饮食或减肥、饮酒、妊娠等诱发。大多数患者发展为慢性肌病症状,包括运动不耐受、肌痛、肌无力、肌肉萎缩。晚发型病人的有机酸尿症常为间歇性,仅在疾病期间和处于分解代谢压力下较为显著。晚发型更多为维生素 B2 反应型。
现有的研究认为,MADD 的临床表现与基因突变类型、位置和环境因素有关。Ⅰ型多为两个无义的纯合突变或无义突变与严重影响mRNA 转录的突变的杂合子,导致致死性结局和先天畸形。Ⅱ型多为影响重要功能的氨基酸或剪切位点的突变,即使很少量的 ETF/ETFDH 活性残余都足以阻止胚胎内的先天畸形的发育。Ⅲ型多为错义突变,突变位置多不在酶活性中心。在更轻症的MADD患者中,ETF/ETFDH 基因型可能受环境因素(如细胞内温度)影响,使得剩余酶活性受到调控而升高。
辅助检查
1.实验室检查 急性发作期可有代谢性酸中毒、低酮症性低血糖、高氨血症。肌肉组织受累可见肌酸激酶升高。肝功能受损导致血浆转氨酶升高、凝血酶原时间(PT)和部分凝血活酶时间(APTT)延长。肾脏小管损伤导致全氨基酸尿。有些 ETFDH 突变导致的轻微的、维生素 B2 反应型表型,生化参数可正常。
2.影像学检查 可见受累器官(肝脏、肾脏)肿大。头颅MRI 可见脑室旁白质脱髓鞘型白质脑病或类似于多发性硬化的白质病变。磁共振波谱分析发现其发作期 Cho/Cr 比值高于正常,其比值可一定程度上反映治疗效果。
3.血浆酰基肉碱检测和新生儿筛查 显示短、中和长链酰基肉碱升高(C4-,C5-,C5DC-,C6-,C8-,C10:1-,C12-,C14-,C14:1-,C16-,C16:1-,C18-,C18:1-,C16-OH-,C16:1-OH-,C18-OH-和 C18:1-OH-酰基肉碱增多)。
4.尿有机酸检测 可见大量戊二酸,因此该病又称为戊二酸血症Ⅱ型或戊二酸尿症Ⅱ型。与戊二酸血症Ⅰ型(GA Ⅰ)不同的是,MADD 不仅有戊二酸的大量排泄,尿有机酸检测还可见乙基丙二酸、3-羟基异戊酸、异戊酰甘氨酸、中链和长链二羧酸的浓度升高。
5.组织活检 肝活检显示脂肪变性,肌活检可见脂质沉积性肌病,心脏、肾脏小管上皮细胞等将脂肪酸作为基本能量来源的组织都可以出现脂质沉积。
6.酶学检测 可通过肝活检或成纤维细胞脂肪酸氧化探针分析进行酶学检测。
7.基因检测 对致病基因(ETFA、ETFB 和 ETFDH)进行测序。
诊断
MADD 是一种可治疗的脂肪酸氧化代谢障碍性疾病,但因其临床表现的高度异质性,给诊断带来难度。
遇阴离子间隙升高型代谢性酸中毒合并乳酸酸中毒、低酮症性低血糖、高氨血症的病例,需疑诊:异戊酸引起的“汗脚”味可作为线索。
诊断试验证实多种酰基辅酶 A 参与的短链、中链、长链脂肪酸,支链氨基酸,肌氨酸代谢产物增多。主要依靠血串联质谱检测和尿气相色谱分析。酶学检测并非必需。确证依靠致病基因的测序。
诊疗流程(图75-1)
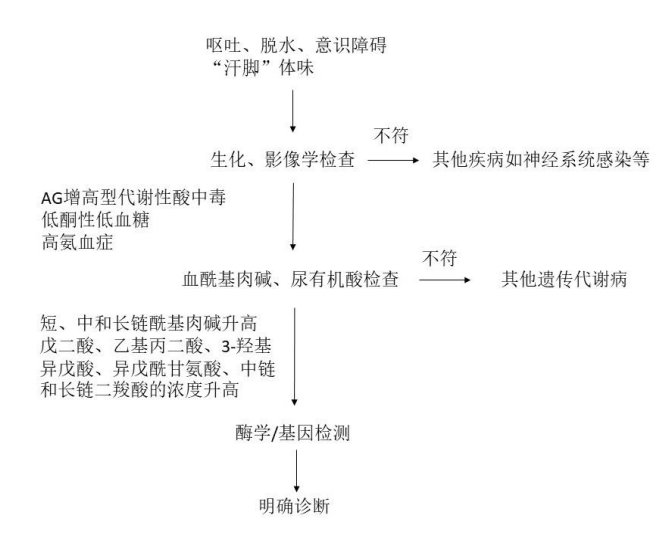
图 75-1 多种酰基辅酶 A 脱氢酶缺乏症(MADD)诊疗流程
参考文献(略)
来源:国家卫生健康委员会《罕见病诊疗指南(2019年版)》
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017