




200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:复旦大学附属华山医院神经内科 衷画画
审核:复旦大学附属华山医院神经内科 罗苏珊
本次世界肌病学会年会(WMS),多项关于LGMD亚型(包括LGMDR4、LGMDR9、LGMDR2和LGMDR7)的研究展示了过去1年从基因治疗临床试验、致病机制解析、功能评估标准化到人工智能辅助预后预测的全链条创新成果。这些研究不仅为理解疾病机制提供了新的视角,更为临床试验设计和未来治疗方案的开发奠定了坚实基础,标志着LGMD精准医疗进入了新的发展阶段。以下是本次会议中LGMD领域的四项重要研究亮点。
LGMDR4基因治疗Ⅲ期研究达到主要终点——Bidridistrogene xeboparvovec显著恢复β-肌聚糖表达
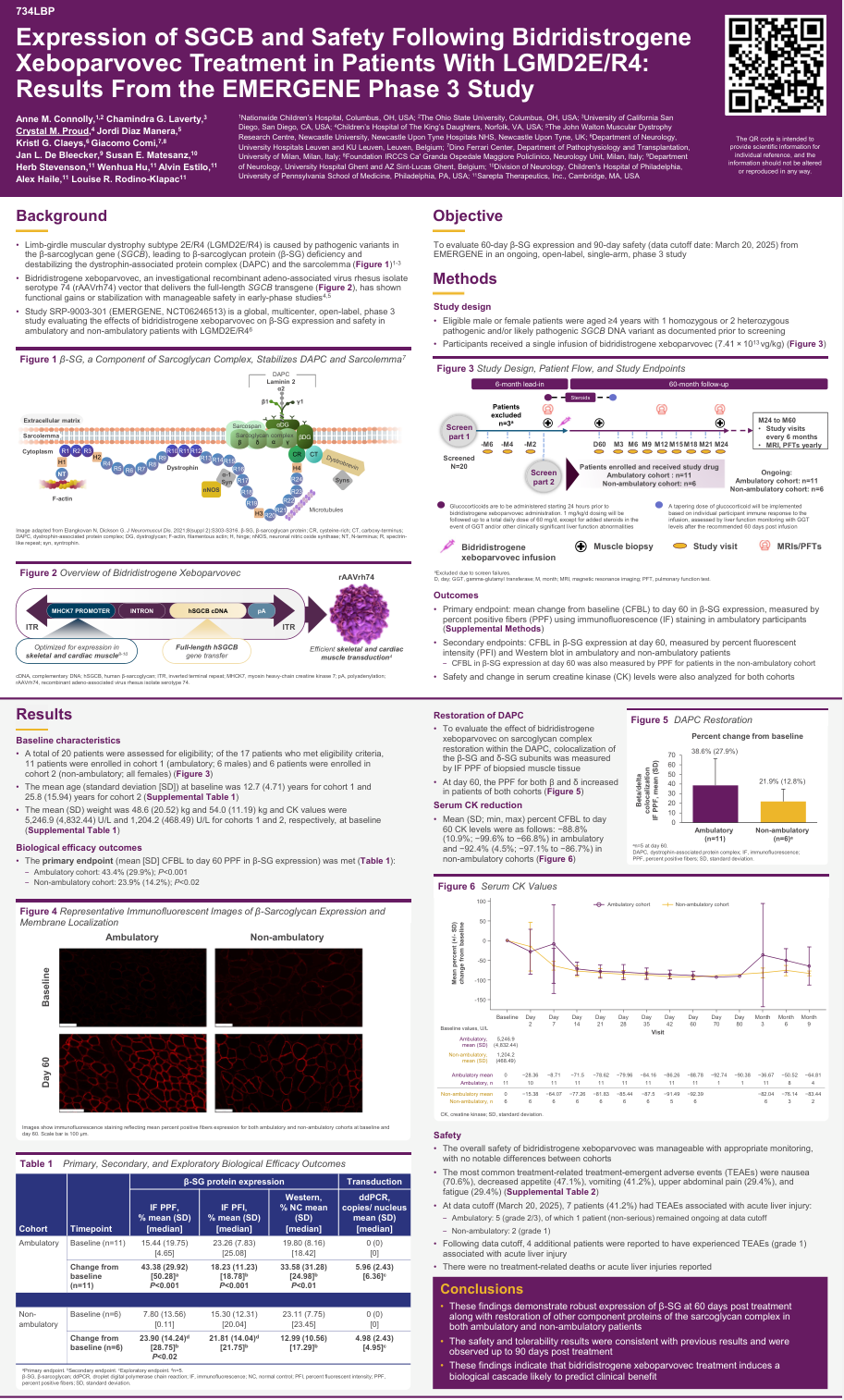
LGMD2E/R4是由β-肌聚糖(SGCB)基因致病性变异引起的进行性肌肉萎缩疾病。在WMS 2025会议上,一项针对LGMDR4的基因治疗临床试验SRP-9003-301(NCT06246513)公布了令人振奋的早期结果,证实了该疗法在恢复蛋白表达和改善生物标志物方面的有效性。
研究设计与关键发现
该研究评估了bidridistrogene xeboparvovec——一种基于rAAVrh74载体递送全长SGCB转基因的在研基因治疗药物。截至2025年3月20日的数据显示,17名年龄≥4岁的患者(行走者11名,非行走者6名)接受了单次剂量为7.41×10¹³ vg/kg的静脉输注治疗。
主要疗效终点成功达成:
行走患者队列:第60天β-肌聚糖(β-SG)表达阳性纤维百分比(PPF)从基线平均增加43.4%(标准差29.9%),P<0.001。
非行走患者队列:β-SG表达平均增加23.9%(标准差14.2%),P<0.02。
更重要的是,研究观察到β-肌聚糖和δ-肌聚糖的共定位增加,这提示肌聚糖复合物得到了恢复——这是维持肌膜稳定性的关键结构。同时,血清肌酸激酶(CK)水平显著下降,行走组和非行走组分别降低了88.8%和92.4%,进一步证实了治疗对肌肉损伤的改善作用。
安全性概况
Bidridistrogene xeboparvovec的整体安全性在适当监测下可管理,两个队列之间无显著差异。最常见的治疗相关不良事件包括:恶心(70.6%)、食欲下降(47.1%)、呕吐(41.2%)、上腹痛(29.4%)、疲劳(29.4%)。7名患者(41.2%)出现与急性肝损伤相关的不良事件(行走组5例[2/3级],非行走组2例[1级])。值得注意的是,目前未报告治疗相关死亡或急性肝衰竭。
该研究为LGMDR4患者提供了首个具有明确蛋白表达恢复证据的基因治疗方案,为后续长期功能改善的评估奠定了坚实基础。随着随访时间的延长,研究团队将继续监测患者的功能结局指标,以全面评估这一创新疗法的临床价值。
在WMS 2025会议上,ATA-001-FKRP研究(NCT05224505)报告了LGMDR9基因治疗的积极中期结果。该项开放标签、多中心剂量递增研究针对确诊为LGMDR9且存在呼吸功能受损的行走患者,评估了单次静脉注射AAV基因治疗药物ATA-100的安全性和有效性。
截至2025年4月,研究共纳入6名患者,分为两个剂量组(9.0E12和2.7E13 vg/Kg),所有患者均为FKRP基因常见L276I突变的纯合子。安全性数据显示,ATA-100总体耐受性良好,治疗后1-3个月出现的无症状转氨酶升高可通过免疫抑制剂有效控制。
令人鼓舞的是,接受首个剂量(9.0E12 vg/Kg)治疗的3名患者在功能和组织学指标上均观察到改善,成功逆转了疾病自然史中预期的功能下降趋势。3个月肌肉活检的生物标志物分析显示,α-肌营养不良聚糖(α-DG)的糖基化水平得到改善,证实了治疗的靶点参与。这一研究为LGMDR9患者带来了新的治疗希望。
AB-1003是一项针对LGMD2I/R9(由FKRP基因突变引起)的AAV介导基因治疗药物。在正在进行的1/2期双盲、随机、安慰剂对照临床试验(NCT05230459)中,首个队列的中期安全性数据展现出令人鼓舞的结果。
研究纳入的5名患者(平均年龄42岁)接受单次静脉输注AB-1003或安慰剂治疗后,随访52周期间未报告剂量限制性毒性或严重不良事件。最常见的不良事件为轻至中度的头痛、跌倒和恶心。3名受试者出现无症状的一过性转氨酶升高,未伴有胆红素水平变化,调整皮质类固醇治疗后恢复至基线水平。基于这些积极的安全性数据,数据安全监察委员会建议研究推进至第二队列。
这项研究的临床前数据已在LGMD小鼠模型中证实,AB-1003能够恢复FKRP酶活性和α-肌营养不良聚糖的功能性糖基化,改善病理和肌肉功能。
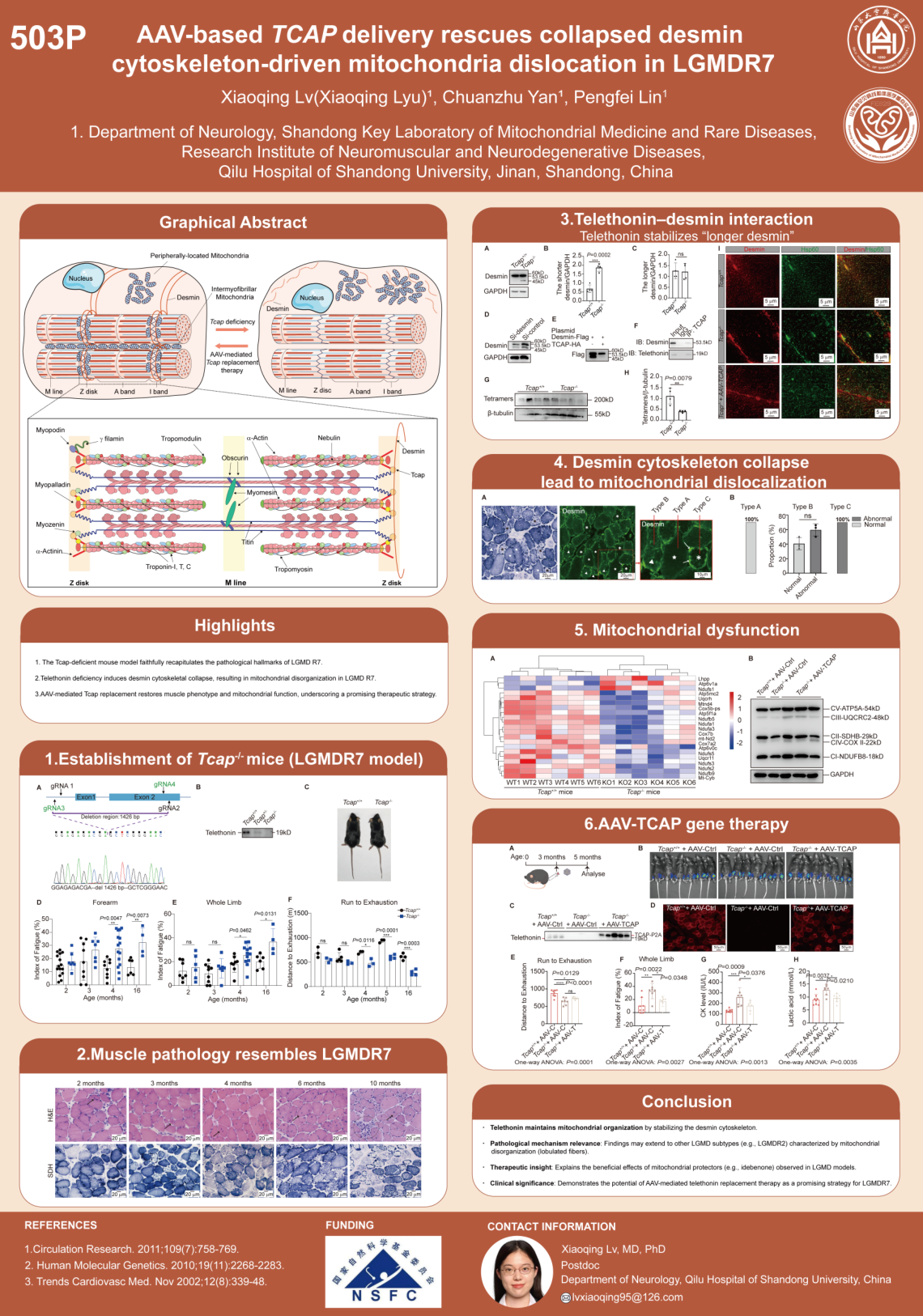
LGMDR7是由TCAP基因突变导致的罕见遗传性疾病,其主要病理特征包括肌纤维大小变异、核中央化和异常的线粒体分布。本研究通过建立Tcap缺失小鼠模型,首次系统阐明了线粒体错位的分子机制。研究团队运用蛋白质组学、免疫荧光、组织病理染色和Western blotting等技术发现,telethonin蛋白通过防止desmin(结蛋白)N端截断来维持其完整性。
Telethonin与desmin结合并共定位于Z盘,对desmin细胞骨架的组织起关键调控作用。当telethonin缺失时,desmin细胞骨架崩溃,导致线粒体网络紊乱和线粒体功能障碍。更重要的是,研究评估了AAV2/9介导的Tcap基因替代治疗效果。肌肉注射AAV后2个月和7个月,Tcap-/-小鼠的肌肉表型、病理、肌酸激酶水平、肌肉MRI、线粒体网络组织和线粒体功能均显著改善。
这项研究不仅揭示了LGMDR7的致病机制,也为AAV介导的基因替代治疗提供了有力的临床前证据。
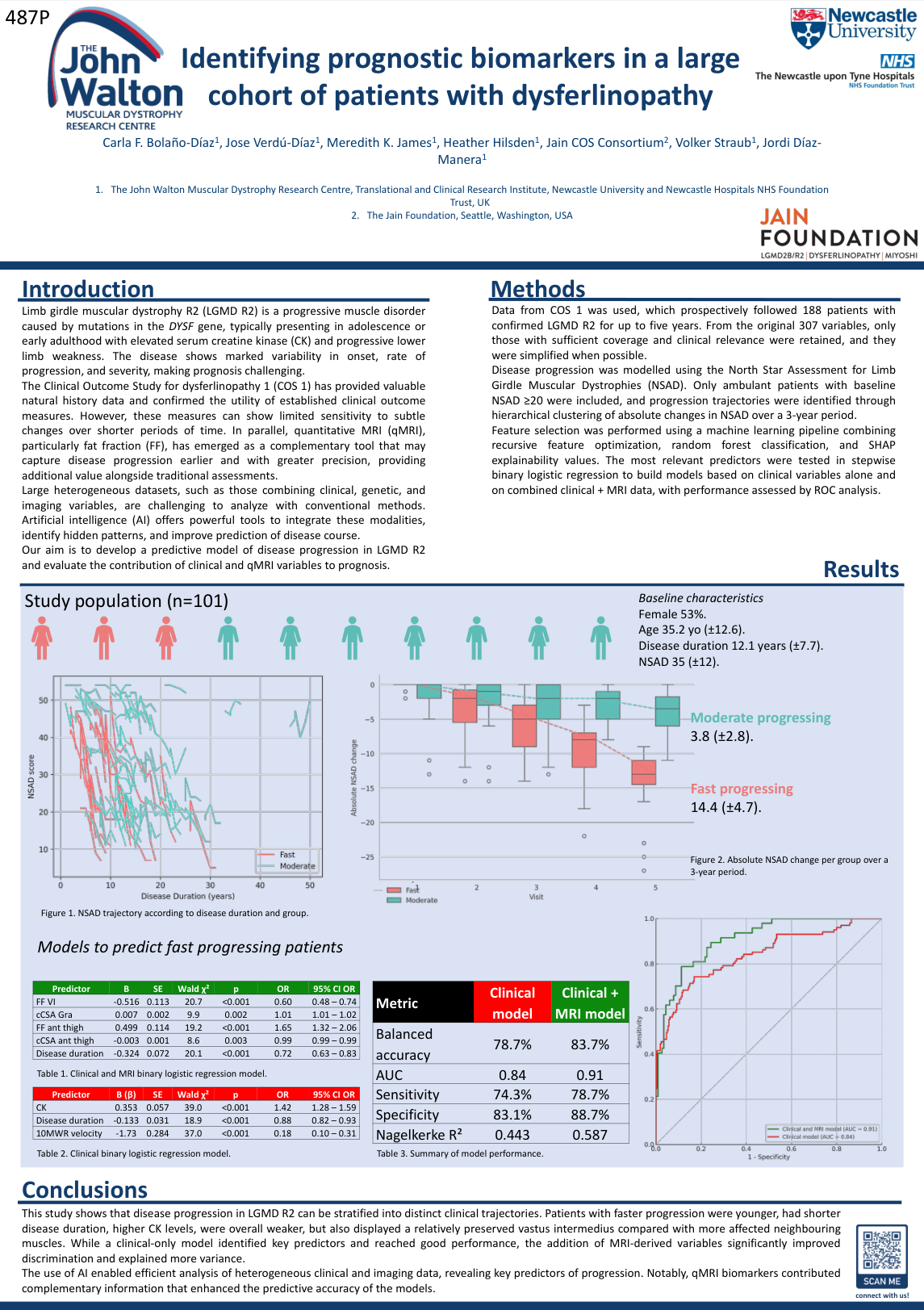
LGMDR2的临床表现复杂多样,疾病严重程度和进展速度存在显著个体差异,这给个体化治疗和临床试验设计带来挑战。本研究首次应用人工智能技术分析了肌联蛋白相关疾病临床结局研究(COS)数据库——迄今最大的LGMD自然史研究数据集。
研究纳入101名NSAD评分≥20分的患者,基于3年内NSAD评分的变化百分比,通过无监督聚类识别出三种不同的进展轨迹:
慢进展组:-4.9%±5.2%
中度进展组:-27.2%±10.4%
快进展组:-39.4%±17.8%
研究团队训练了多个机器学习模型,通过嵌套交叉验证评估,识别慢进展者的准确率达73%,识别快进展者的准确率达81%。SHAP分析揭示了关键预测因子,包括基线NSAD评分、髋内收肌力量(HHM-HAD)以及腓肠肌内侧头、股内侧肌和半腱肌的MRI测量值。
基于这些结果,研究确立了临床阈值:
NSAD评分≥34.5:敏感性78.0%,特异性72.7%
HHM-HAD≥14.7:敏感性88.4%,特异性61.8%
这项研究为临床护理和临床试验中的患者分层提供了有力的AI工具和生物标志物,有望显著改善LGMDR2的精准医疗。
目前LGMDR4与LGMDR9的临床试验均取得积极进展,LGMDR4有望成为最早获治疗的LGMD亚型。中国肢带型肌营养不良患者以R2与R1亚型为主,目前这两种以及其他LGMD亚型均处于临床前研究阶段。此次齐鲁医院团队带来的LGMDR7基因治疗临床前研究为我国领域先河,值得我们共同学习,我们期待其早日走向临床应用。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017