











200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




10月26日,第21届亚洲和大洋洲神经肌病中心年会(AOMC)的议程主要分为四大板块,主要聚焦罕见神经肌肉病、脊髓性肌萎缩症、疑难神经肌肉病及其应对策略、眼-咽远端肌病。

罕见神经肌肉病板块第一位讲者 Khean Jin Goh 教授来自马来西亚大学医学中心,他介绍了马来西亚的家族性转甲状腺素淀粉样变性(hATTR)患者特点。hATTR 是最常见的一种遗传性淀粉样变性疾病,已在葡萄牙、日本、瑞典报道大型家系。在马来西亚,近二十年已有 44 名基因确诊的 hATTR 周围神经病患者,其中 30 位已存在临床症状,起病年龄平均 55.9 岁,起病至诊断平均 3.2 年。其中 86.7%存在感觉伴或不伴运动轴索损害,80%存在自主神经受累。80%人的心超可见心脏受累,1 人存在脑膜受累。在马来西亚,Ala97Ser 最为常见,涵盖了 73%的患者。在马来华侨 hTTR 患者中,超过 80%的患者存在该突变。该突变先前已被报道于中国台湾的一个大型家系,以及泰国和新加坡的家系。常见于中国北方的 Val30Met 在马来华侨家系中也有检出,但占比较少。结合东南亚的历史经济因素,讲者认为该现象可由奠基者效应解释,即既往从中国南部移民至东南亚各国的华侨决定了该基因的高检出率。
第二位讲者是来自美国明尼苏达州罗切斯特市的XinMing SHEN 教授,他分享了“先天性肌无力综合征最新进展”的主题报道。SHEN教授首先分享了合并其他神经疾病的病例,PURA 综合征的特点包括特征是严重智力障碍、癫痫和新生儿期喂养困难、呼吸道和胃肠道疾病、眼睛异常、内分泌缺陷等,临床表现涉及多个系统,以及三个 CMS 合并 PURA 综合征的患儿的临床特点、肌电图检查结果和治疗结局,他们较特异的肌电图结果。随后,他分享了合并 TEFM 突变的 5 个家系,共有 7 个患者,大部分患者都有不同程度的肌肉、大脑、肝脏累及,临床表现广泛,病情较重的患者可有严重的癫痫性脑病、低血糖、乳酸中毒、肝衰竭等,脑部 MRI 可正常也可有非特异性改变,部分患者对沙丁胺醇治疗有反应。动物模型 TEFM 敲除提示神经肌肉接头神经末梢发育受损,为探索发病机制提供线索。
第三个讲者是来自伊朗德黑兰医科大学的 Shahriar Nafissi 教授,他围绕“脂质沉积性肌病”的进行分享,脂质沉积性肌病是一组临床表现高度异质性的遗传性代谢性肌病,以肌纤维内脂肪沉积为主要病理特征,临床特点包括运动不耐受、横纹肌溶解、肌无力等,主要分为 4 个亚型,包括 PCD、MADD、NLSDM、NLSDI。儿童与成人临床表现差异明细,诱因包括发热、压力、劳累。随后通过分享 3 个病例,分别介绍了 PCD、MADD、NLSDM 三个亚型的临床特点、诊断、治疗。总结了伊朗 ETFDH 基因缺陷导致的 MADD 队列,所有患者都有肌肉无力,在接受辅酶 Q10、核黄素、肉碱等治疗后病情好转,其中 84.3%的患者接受治疗后完全康复。最后教授总结了当出现以下情况时应考虑脂质沉积性肌病,包括呼吸受累的肌病,不定时复发,运动不耐受或者横纹肌溶解,CK 轻度升高或者正常,累及上肢和颈肌不对称的肌无力,肌电图提示肌源性损害,也可有自发电位,可通过肌肉活检、酰基肉碱检测、基因检测等手段进行诊断和鉴别诊断。
第四个讲者是来自北京大学第一医院儿科的熊晖教授,她从一个确诊病例开始,介绍了此病的临床特点及进展情况。先天性肌营养不良是一组出生时或出生后 6 个月内出现的原发性、进行性肌病。肌肉活检统一表现出肌营养不良样病变的特征性模式。LAMA2-CMD 典型临床特征是缓慢进行性近端肌无力,肌张力低,大关节挛缩导致自发活动差,进展的脊柱侧弯,癫痫智力正常,频繁的呼吸道感染和呼吸功能不全,肌肉 MRI 显示受累最严重的是臀大肌、大收肌和股二头肌长头,长收肌相对不受累。随后分享了基因型和表型的相关性,LAMA2-CMD 剪接变异的表型通常较轻,LAMA2-LGMD 错义突变更常见,外显子 4 的错义突变更容易出现癫痫,LN 区域的变异更容易出现运动神经病,具有拷贝数变异的患者死亡率可能更高。随后分享了治疗方面的研究,治疗集中 LAMA2 基因治疗、 Laminin α2 同源蛋白替代疗法、炎症抑制、促进肌肉再生等方面。最后教授提到根据中国人群高频的变异位点,已经构建出动物模型,初步揭示了由 Laminin α2 缺乏引起的肌肉萎缩病理变化和白质病变的机制。
来自复旦大学附属儿童医院的王艺教授分享了脊髓性肌萎缩症在中国真实世界的研究数据:中国每年预计增加 850 名患者,估计目前现存 25000 个 SMA 患者。SMA 相关研究自 2019 年后快速发展。中国台湾学者发表了 1 型 SMA 患儿的自然史研究,提到 3 拷贝 1 型 SMA 患儿有更好的生存率,强调早发现早管理的重要性,2020 年中国 SMA 患者的流行病学调查显示,SMA 致病基因的携带率大概是 2%。2023 年中国发表的研究显示展示了诺西那生能够改善 SMA 患儿的运动功能,中国临床医生取得的研究成果正在向全世界展示。
Sophelia教授分享的主题是 SMA 治疗-未满足的需求”。SMA 的分型与 SMN 的拷贝数相关,SMN2 拷贝数越多,病情越轻,其中 1 型患者病情快速进展,未治疗的情况下大多 2 岁内死亡,目前共有三个药物被 FDA 批准,包括诺西那生、利司朴兰和 Zolgensma。Sophelia教授详细介绍了自己团队在 SMA 的治疗的研究,研究结果显示,不同运动功能状态的 1 型和 2 型患者得到了不同程度的改善,1 型和 2 型患者观察到明细的脊柱侧弯进展,尤其是在 5 岁和 10 岁之间才接受治疗的患者。HFMSE 评分改善越明显,脊柱侧弯进展的速率越慢,虽然所有亚组接受治疗后运动功都能改善,但是在没有脊柱侧弯的患者中改善得更明显,因而提出诺西那生治疗并不能预防已出现肌肉无力的 2 型 SMA 患者的关节并发症的观点。随后Sophelia教授分享了其他SMA患者在骨健康领域的研究,包括中国在 2 型和 3 型 SMA 患者骨密度研究和影响因素。
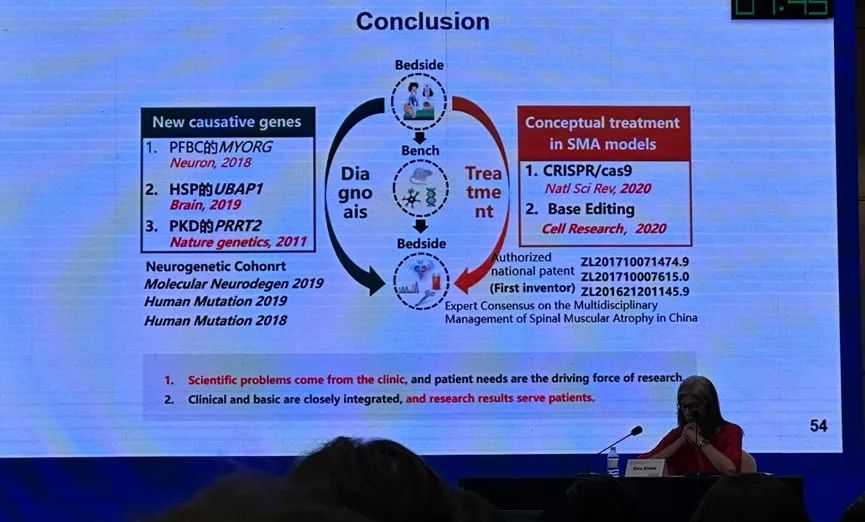
疑难神经肌肉病及其应对策略板块的第一位讲者是福建医科大学第一附属医院的陈万金教授,他讲授了神经肌肉病致病基因发现策略及相应基因治疗的探索。陈教授从一例 OPDM 患者的基因诊断出发,着重介绍了基因非编码区在神经肌肉病诊治中的价值。编码区(外显子)占基因组 1%,携带 80%遗传信息,而非编码区(内含子)在基因组占比远大于编码区,携带遗传信息较少,但对其他单基因及多基因遗传病具有提示作用。除探索新的致病基因以外,还应着重寻找致病基因的可变碱基动态突变。此外,基于非编码区基因,靶向内含子利用反义寡核苷酸、CRISPR/Cas9 进行干预,在遗传性神经肌肉病的治疗中具有较大前景。陈教授还分享了团队发现遗传性神经肌肉病新致病基因和 VNTR 变异的经验,并介绍了自主设计 ASO 和 CRISPR/Cas9 治疗模型鼠的进展。
第二位讲者是来自首尔国立大学的 Jong Hee Chae 教授,她讲授了在疑难神经肌肉病中基因检测的选择策略。神经肌肉病疾病谱多样,具有临床异质性。遗传性神经肌肉接头及肌肉疾病的患病率约为 2~6/10000 人,在基因组学飞速发展的时代,基因检测已为临床诊疗提供了大量帮助。精准分子诊断有助于精准治疗,避免不必要的检查和诊断性治疗药物的副作用,还可助力遗传咨询。然而,如何决策基因检测的使用,以及如何选择合适的检测手段,是临床医生需要思考的问题。讲者从既往接诊的几例疑难患者的诊治过程出发,强调了首先应进行准确充分的临床评估,再考虑行必要的基因检测,还应基于疾病表型,选择合适的基因检测策略。
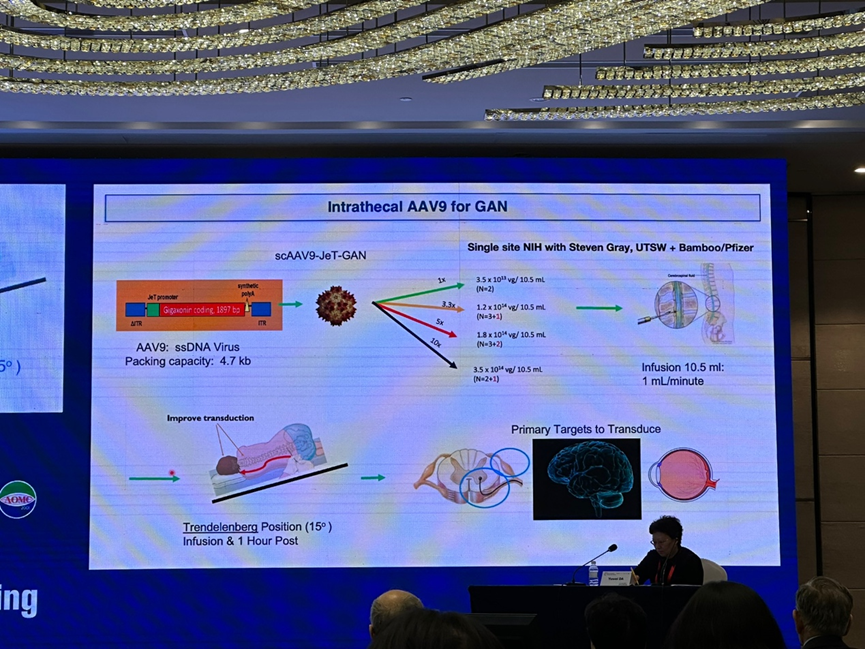
第三位讲者是来自美国国家神经疾病及卒中中心的 Carsten Bönnemann 教授,他详细介绍了腺病毒(AAV)基因治疗在神经肌肉病患儿治疗中的机遇与挑战。基因治疗能够直接从发病机制进行干预,能够精准治疗疾病,尤其是特别罕见的疾病,具有显著的优越性。现今人们对于 AAV 介导的基因治疗已有成熟的生产流程,并能根据不同的 AAV 对于人体组织的亲和性和特定的给药方式进行设计,以确保将药物精准递送到神经系统的各个组成部分。Bönnemann 教授结合现已开展于 SMA、巨轴索周围神经病(GAN)的临床研究,呈现了 AAV 基因治疗的显著疗效,及遗传性神经肌肉病及早进行诊断和基因治疗必要性。除有效性以外,还需要关注 AAV 治疗的免疫安全性及免疫毒性,如 AAV 中和抗体、抗转基因免疫等,该类因素都可能影响远期疗效和患者恢复过程,对 AAV 基因治疗的应用产生较大挑战,仍需深入研究。
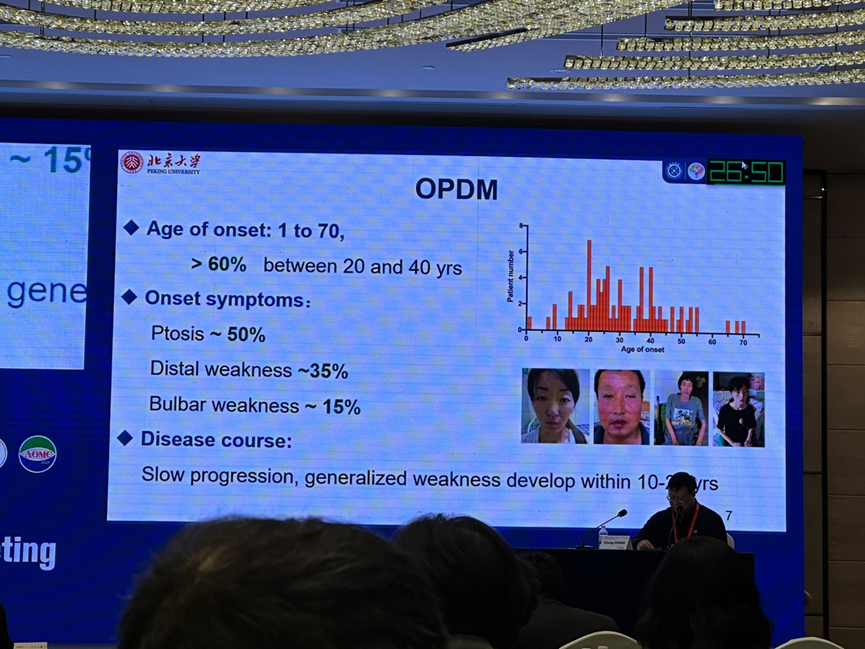
眼-咽远端肌病(OPDM)板块的第一位讲者是来自北京大学第一医院的王朝霞教授,她介绍了团队发现 3 个 OPDM3 致病基因的过程。OPDM 是一类罕见的成人起病的肌肉疾病,临床以进行性眼外肌、面肌、咽喉肌及远端肢体肌无力为特征性表现。OPDM 的典型肌肉病理表现为镶边空泡和核内包涵体。LRP12 是第一个被发现的 OPDM 的致病基因,王教授团队在国内率先报道了 GIPC1 基因突变导致 OPDM 的家系,而后又通过不断探索,发现了 NOTCH2NLC、RILPL1 作为 OPDM 的致病基因。OPDM 是一类重复扩增相关神经肌肉病,CCG 拷贝数的增加,与发病年龄较早具有相关性。但在各致病基因之间,临床表型和肌肉 MR 表现都较为接近。OPDM 可合并其他疾病,其中 NOTCH2NLC 的系统性损害较为显著,而新发现的 RILPL1 基因突变相关 OPDM 的合并表现特点尚不明确。
第二位讲者是来自复旦大学附属华山医院的奚剑英教授,她分享了 OPDM 的临床表现谱系特点。在中国,OPDM 患者以 GIPC1 基因突变为主,同时也存在 LRP12、NOTCH2NLC、RILPL1 突变的患者。这些患者的 OPDM 典型表现大抵相似,但也可在典型表现以外叠加其他疾病的临床特点。OPDM 根据致病基因的不同分为 4 型,其中 OPDM1 为 LRP12 基因突变导致,除典型的 OPDM 表现以外,还具有神经元核内包涵体病(NIID)、ALS 或 PMA 样表型。OPDM2 为 GIPC1 基因突变导致,可伴有听力丧失、周围神经损害、运动障碍疾病等。OPDM3 为 NOTCH2NLC 基因突变所致,可存在肾脏损害,眼底及 OCT 检查可有异常表现。OPDM4 由 RILPL1 基因突变导致,患者的起病年龄更早,上睑下垂更为显著。奚剑英教授还介绍了团队近期发现的 LOC642361/NUTM2B-AS1 基因突变导致的 OPDM 病例。
第三位讲者是来自南昌大学附属医院的洪道俊教授,他详细阐述了 NOTCH2NLC 基因突变相关 OPDM 的病理特点和发病机制。NOTCH2NLC 基因突变相关 OPDM 的典型肌肉病理表现为镶边空泡和核内包涵体,而该疾病发生的遗传学机制与 CGG 重复扩增所致的蛋白质毒性和 RNA 毒性相关。既往在 NIID 中发现,AUG 可介导 CGG 翻译产生 polyG 的堆积,通过蛋白质毒性造成疾病发生。在肌肉病理中,洪教授团队发现 RNA 团簇与核内包涵体共定位,提示核内包涵体的 RNA 毒性,进而通过设计 AAV 小鼠模型及果蝇模型,验证了包涵体中的 RNA 和蛋白质毒性。洪教授团队还发现 polyG 与线粒体共定位,而 IDB 药物治疗,能够部分缓解疾病受累。进而明确了在 NOTCH2NLC 基因突变相关 OPDM 中,gain-of-function 为疾病发生的主要机制。OPDM 的各个分型虽致病基因不同,但疾病表型和肌肉病理具有类似表现,各型的遗传学机制是否存在同质性,还需进一步研究明确。
来源:华山罕见病(供稿:胡嘉年|王宁宁|罗苏珊)

查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017