


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过





近年来肝癌的发病率有所上升,尤其是肝细胞癌(HCC),而肝癌干细胞(liver cancer stem cells,LCSC)在HCC的发生发展中起着关键作用[1]。LCSC又称为肝癌起动细胞,是一类能自我更新并分化形成肿瘤的细胞,它引发HCC并且是导致其复发的根源[2]。
肿瘤微环境(tumor microenvironment, TME)是指肿瘤周围的微环境,由非细胞成分和细胞成分组成,前者有细胞外基质(ECM)、细胞外调控分子、细胞外囊泡(extracellular vesicles,EV)和其他环境因素,后者有内皮细胞、免疫细胞、癌相关成纤维细胞(cancer-associated fibroblasts,CAF)、干细胞和其他细胞;肝脏中的TME除了上述成分外还包括LCSC、肝窦内皮细胞(liver sinusoidal endothelial cells,LSEC)和肝星状细胞(HSC)。有研究[3]表明TME中的这些成分可以改变LCSC的可塑性,促进其表型和功能的改变,导致HCC的发生、转移和耐药。
在此,这篇文章介绍TME中的主要成分:ECM、细胞外调控分子、EV、血管内皮细胞、免疫细胞、CAF和其他细胞,如何协同上皮-间质转化(epithelial-mesenchymal transition,EMT)、肿瘤新生血管网络,激活STAT3、TGFβ、Hedgehog、PI3K/AKT等信号通路,调节LCSC的生成、增殖、自我更新、分化、免疫逃逸、干性和化学抗性,影响肝癌细胞的发生、发展。此外,本文还描述了TME中的主要成分及LCSC相关联的分子靶点和治疗的方法或药物,并对一部分治疗方法的局限性做出讨论。最后,本文在微环境各成分和LCSC的基础上对未来的肝癌分子靶向联合治疗上提出了展望。

ECM是CAF等细胞产生并分泌到细胞外空间的非细胞形态物质,呈复杂而精密的网络结构,按其组成和结构的不同,分为间质基质、细胞周围基质和基底膜。间质基质围绕细胞,有胶原蛋白、弹性蛋白、蛋白聚糖、非胶原糖蛋白,而细胞周围基质与细胞紧密接触[4]。(1)胶原蛋白是动物体内高度特化的纤维蛋白家族,是人体内含量最丰富的蛋白质,主要由成纤维细胞分泌,至少有28种类型:Ⅰ型主要构成胶原纤维,Ⅲ型主要构成网状纤维,Ⅳ型合成基底膜[4];(2)弹性蛋白主要构成弹性纤维,是ECM中弹性纤维网络的主要成分;(3)蛋白聚糖,也称蛋白多糖,是氨基聚糖与核心蛋白质以共价键结合而成的聚合体,成分主要是透明质酸(hyaluronic acid,HA);(4)非胶原糖蛋白,主要是纤连蛋白(fibronectin,FN)和层粘连蛋白。
ECM能影响LCSC的恶性标志物的表达、细胞间通讯,促进HCC的发生和发展。据报道[5-6],ECM的重塑增加了LCSC相关标志物(如CD44、CD133)的表达,促进HCC的增殖、侵袭和化疗抗性;此外,HA与CD44相互作用,促进LCSC增殖扩张,影响HCC的发生[6];Kaplan等[7]发现FN受体(整合素α4β1)的特异性抗体可以中断LCSC和微环境之间的相互作用,抑制HCC的转移。其相关作用机制汇总见表 1。
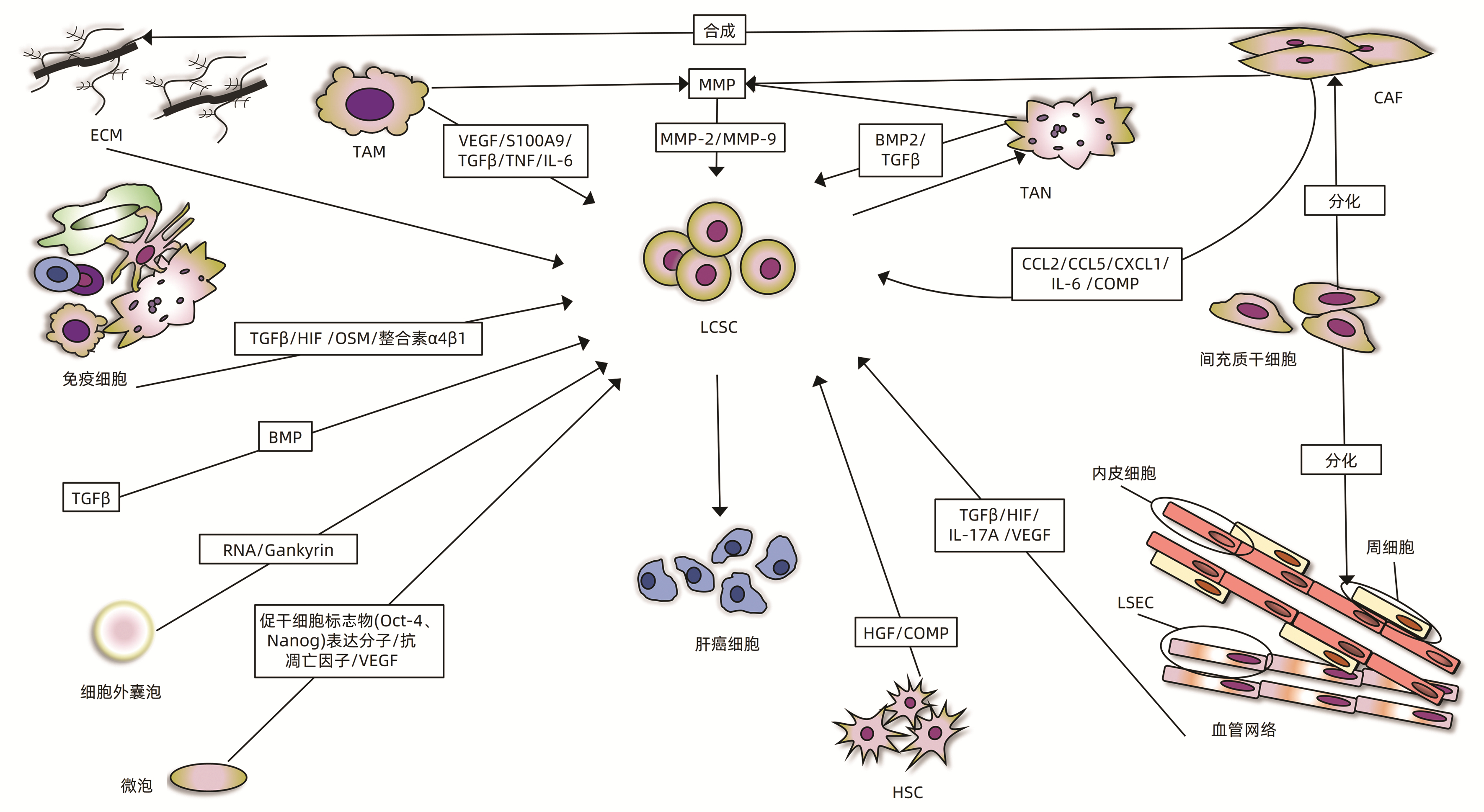
图 1 肿瘤微环境与肝癌干细胞相互作用而影响肝细胞癌的发生和发展
注:COMP,软骨寡聚基质蛋白。
1.2 细胞外调控分子
TME中的细胞分泌多种调节分子,如多种细胞分泌的HIF、TGFβ,免疫细胞分泌的制瘤素M(oncostatin- M,OSM)和其他细胞外调控分子,这些成分与重塑的ECM、EMT和肿瘤新生血管网络相互协同,激活Notch、TGFβ、STAT3信号通路,增强LCSC的干性和化学抗性,对肝癌的进展产生积极影响(图 1)。
HIF是一种介导细胞缺氧反应的核蛋白,具有转录活性和相当广泛的靶基因。HIF协同ECM重塑、肿瘤新生血管网络和EMT,参与LCSC的生成、维持和化学抗性,从而介导肝癌的发生和耐药[2, 8-9]。例如,有研究[10-11]发现HIF-1α驱动EMT参与人HCC细胞干样特征的获得:LCSC标志物表达增加、集落形成能力和球体形成能力增强,促进HCC的发生;在HCC中,HIF-1α/Notch信号通路对膜联蛋白A3(annexin A3,ANXA3)维持LCSC活性至关重要[6];Bort等[10]发现AMPK通过上调HIF-1α诱导LCSC重编程,恢复其对索拉非尼的敏感性;Cheng等[12]发现阻断Akt/HIF-1α/PDGF自分泌环可减弱缺氧条件下LCSC和肝癌细胞的化学抗性;HCC中,HIF-1α的转录激活可以对LCSC起支持作用,进而诱发HCC的进展和治疗抵抗[13]。其相关作用机制汇总见表 1。
TGFβ是一种调节细胞生长、分化的多功能细胞因子,在TME中高度表达。TGFβ通过改变LCSC干性来对HCC的发生、侵袭、转移发挥促进作用,如骨形态发生蛋白(bone morphogenetic protein,BMP)是TGFβ家族中的一个亚群,有研究[12]表明给予大量外源性的BMP4能够促进CD133+LCSC的分化,抑制它的自我更新、化学抗性和致瘤性;BMP9和EMT通过激活TGFβ信号通路,增强DNA结合蛋白1抑制剂的表达,减少E-钙黏蛋白来促进上皮细胞黏附分子(epithelial cell adhesion molecule,EPCAM)的表达,减少细胞黏附,从而诱导LCSC发生恶性表型转化,参与HCC细胞的侵袭和转移过程[14-17](图 1)。其相关作用机制汇总见表 1。
OSM是一种IL-6家族成员, 主要由免疫细胞分泌,如活化的T淋巴细胞、活化的单核-巨噬细胞等,研究[12, 18]发现OSM可以通过激活STAT3信号通路使LCSC分化为癌细胞,启动肝癌的发生(图 1)。MMP是一类由CAF、TAM、TAN等分泌的Zn2+和Ca2+依赖的蛋白酶,有胶原酶、明胶酶、基质溶解素、弹性蛋白酶等类型,MMP-2和MMP-9表达的增加,增强LCSC的自我更新和化学抗性,影响HCC治疗的效果[19](图 1);化合物4, 4′-健黑麦铜酸D通过下调MMP-9和金属蛋白酶-1的拮抗组织抑制剂来抑制LCSC,从而阻止了HCC的转移[20]。OSM和MMP相关作用机制汇总见表 1。
EV是一种双层磷脂膜囊泡,内含RNA、蛋白质、DNA和脂质等,参与细胞间的物质转运和信息交流,它可被几乎所有类型的细胞释放,EV根据其大小和细胞室的起源,可分为外泌体、微泡(microvesicles,MV)和凋亡小体[21]。外泌体是EV中较小的一种,平均直径约为100 nm,癌细胞来源的外泌体通过转移miR-125b、miR-130、miR-155和Kras mRNA参与CSC重编程和肿瘤形成[22-24](表 1);而miR-21和miR-9-3p可作为肝癌的诊断标准和治疗靶点[25](表 1)。MV又称细胞外微泡,脱落微泡或微粒,直径100~1000 nm,较外泌体大。由于凋亡小体一般会被巨噬细胞吞噬消除,本文不做过多叙述。
EV参与LCSC微环境的改变,是HCC发生和发展的强有力的刺激因素。EV富含miRNA-181时,可通过激活Wnt/β-catenin信号通路,增强β- catenin活性,抑制LCSC的分化和促进EpCAM+ LCSC的自我更新,同时增加miRNA-130b、lncTCF7的表达,诱导肝癌的发生和化疗耐药[26-27];另外,miRNA-181表达的增加,能增强LCSC的恶性表型和功能特点,体外研究[26]表明通过递送miRNA-181特异性抑制剂来下调miRNA-181的表达,从而抑制LCSC的干性基因表达和自我更新。当EV富含Gankyrin(也称为p28GANK、p28或PSMD10)时,一种重要的癌蛋白,激活PI3K/AKT信号通路,调节LCSC在肝癌细胞的增殖、凋亡和化疗耐药中的作用[16, 28](图 1)。EV相关作用机制及功能汇总见表 1。
缺氧、pH变化、细胞代谢产物污染导致的氧化应激和自噬通过LCSC对肝癌的发生发展起着支持作用(表 1)。自噬是一种通过清理受损细胞器来适应环境的分解代谢途径,Zhao等[29]发现在体外培养Huh7细胞时,自噬标志物LC3-Ⅱ表达增加的细胞,Western Blotting检测出其干性相关基因Nanog、Sox2和Oct4的表达也增加,以上观察结果支持了自噬在维持LCSC干性上发挥关键作用。
总之,ECM与其他TME中的非细胞成分作为细胞通讯的基础,与LCSC相互作用,产生有利于肝癌细胞更好生存的微环境,从而出现HCC治疗困难的局面。
TME中的细胞成分分为血管内皮细胞、免疫细胞、CAF和其他细胞。过往研究[4, 8]表明,这些细胞直接或间接参与ECM重塑,EMT和肿瘤新生血管网络形成,并且激活LCSC相关干性通路,促其免疫逃逸,改变其恶性表型和功能,影响HCC的形成过程。
血管内皮细胞通常是指衬于心、血管和淋巴管内表面的单层扁平上皮,它形成血管的内壁。HCC是典型的多血管实体瘤,有着异常丰富的血管网络,其组成有内皮细胞、周细胞和LSEC(图 1)。血管网络的形成有依赖内皮细胞型和非依赖内皮细胞型,(1)依赖内皮细胞型:肝癌细胞可以直接募集内皮祖细胞到达肿瘤部位分化为内皮细胞,形成新生血管;或者肝癌组织中现有的内皮细胞发芽形成新生小血管[30]。(2)不依赖内皮细胞型:血管生成拟态(vasculogenic mimicry,VM)是一种不依赖血管生成的肿瘤微循环模型,由肿瘤细胞本身形成类似血管并具有基底膜的小管状结构,可与血管相通,在HCC中,CD44、Oct4和EMT相关基因表达的上调,能促进VM的形成,这是在没有内皮细胞的情况下肝癌细胞依旧拥有良好血液供应的原因之一[6]。
肿瘤新生的血管网络通过分泌多种因子促进LCSC的恶性改变。2022年的一项研究[31]表明淋巴管内皮细胞分泌IL-17A上调LCSC中PD-L1而促进HCC发生;内皮细胞分泌HIF-1α和血管内皮生长因子(vascular endothelial growth factor,VEGF),增加LOXL2的表达,诱导EMT和VM的形成并协同ECM重塑,增强LCSC的维持和自我更新,参与HCC的发生和转移[5, 32-33];内皮细胞还分泌HIF和TGFβ通过LCSC发挥促瘤作用,具体作用机制上文已提及(图 1)。内皮细胞与血管网络的相关作用机制汇总见表 1。
免疫细胞又称炎细胞或白细胞,是指参与机体免疫应答的各种细胞的总称,包括淋巴细胞、单核-巨噬细胞等,起到免疫防御、免疫监视和免疫自稳的作用。各种肝损伤、感染激活免疫细胞,使微环境出现高炎状态,它和新生的肿瘤血管一起,诱导肝祖细胞或者成熟的肝细胞获得干性,转化为LCSC,并促其逃避免疫监视,参与肝癌的发生、侵袭和转移[12, 34-35]。例如,TAM可以通过释放VEGF、MMP、分泌蛋白S100A9、TGFβ和肿瘤坏死因子(TNF),激活IL-6/STAT3信号通路,与肿瘤新生血管一起来改变LCSC表型和功能,引起HCC,并增强肿瘤细胞侵袭能力[2, 6, 12, 36](图 1)。TAN分泌BMP2和TGFβ来诱导LCSC的产生,而LCSC的增加反过来又招募了更多TAN,从而在HCC中形成了一个正反馈环[37]。正常情况下树突状细胞和T淋巴细胞对LCSC的维持与免疫逃逸至关重要,而经ANXA3转染的树突状细胞可有效激活T淋巴细胞,导致LCSC的特异性杀伤,使其免疫逃逸失败[6, 12](图 1)。免疫细胞相关内容汇总见表 1。
CAF又称肿瘤相关成纤维细胞,是TME中的成纤维细胞亚群,呈持续活化状态,其来源有成纤维细胞、经历EMT转化的HCC细胞、骨髓干细胞等。微环境中CAF分泌多种因子介导ECM重塑,激活TGFβ、Hedgehog、STAT3等信号通路,来促进LCSC恶性表型的转化和肝癌侵袭和转移。许多研究[6]表明,CAF可以通过合成胶原蛋白参与ECM的重塑并分泌CCL2、CCL5、CXCL1、IL-6等细胞因子激活TGFβ、Hedgehog、STAT3和Notch信号通路,增强LCSC干性;此外,它分泌的COMP激活MEK/ERK和PI3K/AKT信号通路驱动EMT,激活LCSC恶性转化。Zhao等[29]发现在CAF的条件培养基里培养Huh7细胞时,Huh7细胞成球增多,干性相关基因表达增加,表明了CAF诱导HCC细胞表达干样特性,使得非LCSC癌细胞向LCSC转化,重新进入干细胞状态,导致HCC细胞的侵袭和转移能力的增加(图 1)。其相关作用机制汇总见表 1。
间充质干细胞从骨髓中被募集,分化为CAF、周细胞、脂肪细胞,起到促血管生成,促侵袭,支持LCSC的作用,从而影响肝癌的发生发展[38-39];LSEC和HSC通过调控免疫反应,改变LCSC干性特点来发挥促瘤作用[40];HSC通过分泌肝细胞生长因子、COMP,在LCSC促进HCC的发生上发挥作用[6, 41](图 1)。总体上,应进行更深一步的研究来确定TME的细胞成分与LCSC的具体的相互作用机制,以及它们对肝癌的影响。
上文已经提及TME和LCSC相互作用的机制,这部分根据这些机制总结了两者相关联的一些分子靶点及治疗方法或药物(表 2)。
表 2 肿瘤微环境各成分和LCSC相关联的分子靶点、治疗方法或药物
Table 2. Molecular targets, therapeutic methods or drugs associated with various components of the tumor microenvironment and the LCSC
分子靶点 | 治疗方法或药物 | TME各成分靶点对LCSC的作用 |
ECM | 胶原蛋白参与ECM重塑导致LCSC干性标志物CD44、CD133表达的增加;FN影响LCSC细胞间通讯[5-7, 42-43] | |
胶原蛋白 | 吡格列酮 | |
CD44 | 抗CD44抗体; miR-199a-3p | |
CD133 | MV-141.7;MV-AC133 | |
FNR | 抗整合素α4β1抗体 | |
HIF | 内皮细胞分泌HIF-1α对LCSC的恶性改变起支持作用;Akt/HIF-1α/PDGF自分泌环增强LCSC的化学抗性[2, 5, 8-13, 44-45] | |
HIF-1α | HDAC6特异性抑制剂 | |
PDGFR | 索拉非尼; 仑伐替尼; 瑞戈非尼 | |
TGFβ | siRNA; 吡非尼酮; 氟非尼酮 | TGFβ和BMP导致LCSC干性标志物CD133、EPCAM表达的增加[12, 14-17, 42, 46-48] |
BMP | DMH1 | |
EpCAM | VB4-845;Edrecolomab; RNAi | |
其他细胞外调控分子 | IL-6、MMP和TIMP对LCSC的恶性改变起支持作用[12, 18-20, 42] | |
IL-6 | 阿司匹林 | |
MMP | FR(EtOH) | |
TIMP | 常山酮 | |
EV | 载药RBC-EV | EV、Wnt/β-catenin信号和lncTCF7对LCSC的恶性改变起支持作用[22-27, 42-43] |
Wnt/β-catenin | siRNA | |
lncTCF7 | - | |
其他环境因素 | 缺氧、氧化应激和自噬LCSC的恶性改变起支持作用[29, 42-43, 49-50] | |
缺氧 | Evofosfamide (TH-302);DS-PLGA | |
氧化应激 | Oroxylin A; 甲基阿魏酸 | |
自噬 | 3-甲基腺嘌呤; 巴弗洛霉素A1 | |
内皮细胞/血管 | 阿替利珠单抗 | 内皮细胞通过PD-L1、HIF-1α、VEGF和LOXL2对LCSC的恶性改变起支持作用[5, 31-33, 42, 44-47] |
PD-L1 | HDAC6特异性抑制剂 | |
HIF-1α | ||
VEGF | 贝伐珠单抗 | |
VEGFR | 索拉非尼; 仑伐替尼; 瑞戈非尼; 卡博替尼 | |
LOXL2 | 辛妥珠单抗 | |
TGFβ | siRNA; 吡非尼酮; 氟非尼酮 | |
免疫细胞 | TAM通过分泌VEGF、MMP、TGFβ和TNF激活IL-6/STAT3信号通路对LCSC的恶性改变起支持作用;而TAN通过BMP2和TGFβ对LCSC的恶性改变起支持作用[2, 6, 12, 36-37, 42, 45-49] | |
TAM | 索拉非尼 | |
VEGF | 贝伐珠单抗 | |
MMP | FR(EtOH) | |
TGFβ | siRNA; 吡非尼酮; 氟非尼酮 | |
TNF | 阿司匹林 | |
IL-6 | 阿司匹林 | |
STAT | 利匹韦林 | |
BMP | DMH1 | |
CAF | CAF分泌胶原蛋白、TGFβ、CCL2、CCL5和IL6参激活TGFβ和STAT3信号通路对LCSC的恶性改变起支持作用[6, 42, 46-47, 51] | |
TGFβ | 吡非尼酮; 氟非尼酮 | |
STAT | 利匹韦林 | |
胶原蛋白 | 吡格列酮 | |
CCR2/5 | Cenicriviroc | |
CCR5 | Met-CCL5 | |
IL-6 | 阿司匹林 | |
注:FNR,纤连蛋白受体;HIF,缺氧诱导因子;PDGF/PDGFR,血小板衍生生长因子及受体;RNAi,RNA干扰;siRNA,小干扰RNA;TIMP,金属蛋白酶组织抑制剂;RBC-EV,红细胞释放的细胞外囊泡;VEGF /VEGFR,血管内皮生长因子及受体;CCR2/5,CCL2/5的受体。 | ||
然而,以上治疗都有其局限性。其中免疫治疗有如下三个问题需要进一步探讨:(1)免疫细胞主要通过诱导LCSC进行肝源性分化,但如何诱导LCSC分化为正常肝组织而不是肝癌组织尚未可知;(2)由于LCSC的异质性,免疫治疗时如何很好地靶向LCSC仍是一个问题;(3)其他干细胞或者成熟肝细胞转化而来的LCSC带有其亲代相关表面标志物,体外靶向LCSC的同时如何避免损伤到亲代细胞和其他正常的细胞。此外,VM的存在是传统抗血管药物治疗肝癌不成功的原因之一,那么加大VM靶向疗法的开发,或者说把传统疗法与VM靶向疗法相结合,未尝不是一个新的方向。最后,载药RBC-EV,由于EV是由细胞本身发芽而成,难免带有部分亲代细胞膜标记的特有的信息。例如,给癌症患者使用血小板源性EV治疗时,它会向肿瘤细胞转移并融合来自血小板的多个内皮靶向黏附受体,使得肿瘤细胞的转移性增强[52];另外,在极少数情况下,体外扩增EV会得到促进肿瘤细胞启动的EV,有一定的疾病靶向和生物安全问题[52];当MV富含促进干细胞标志物(Oct4、Nanog)表达的分子、抗凋亡因子、VEGF等分子时,会使正常肝细胞增生,提高其存活率;同时,也抑制肝癌细胞的凋亡和免疫反应,有增加化疗抵抗性,形成畸胎瘤等不好的风险[52](图 1)。
本文旨在探讨TME中的细胞和非细胞成分与LCSC的相互作用机制,并对肝癌细胞的发生、侵袭、转移和耐药产生重要影响作用,希望为针对TME和LCSC相关联的分子靶点治疗HCC提供相应的线索,有助于更好的开发HCC靶向治疗方法。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017