


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:东部战区总医院 国家肾脏疾病临床医学研究中心 左科 成水芹 陈文萃 安玉
通信作者:左科
病史摘要:患者青年男性,半年前劳累后出现双下肢浮肿,后检查发现血压升高,大量蛋白尿伴血肌酐升高,合并轻度增生性贫血,高密度脂蛋白明显降低,无关节痛、皮疹等其他症状,自身抗体检查未见异常,父亲及爷爷年轻时均出现过浮肿,父母近亲结婚(姑表关系)。
症状体征:双下肢浮肿、查体可见角膜混浊,伴血压升高。
诊断方法:肾活检病理提示肾小球基底膜空泡状改变,个别空泡内见嗜锇性物质,通过基因检测证实患者卵磷脂胆固醇酰基转移酶(LCAT)相关基因纯合突变,导致LCAT缺如。
治疗方法:口服贝那普利护肾,降脂药物非诺贝特及阿昔莫司,并给予双重血浆滤过置换治疗,总计输注血浆3740ml。
临床转归:患者出院时血肌酐及血白蛋白升高恢复至正常,仍有轻度贫血。
关键词:卵磷脂胆固醇酰基转移酶缺乏症;家族性LCAT缺乏症;鱼眼病
引 言
卵磷脂胆固醇酰基转移酶(Lecithin-Cholesterol Acyl Transferase, LCAT)缺乏症是一种由基因突变引起的罕见的常染色体隐性遗传疾病, 根据临床表现形式和LCAT活性缺乏程度分为两类:家族性 LCAT缺乏症(Familial LCAT Deficiency,FLD)和鱼眼病(Fish Eye Disease,FED),前者临床表现有非免疫性溶血性贫血、眼角膜浑浊和肾脏受累,后者主要临床表现为角膜浑浊。这里我们报道1例典型FLD患者的诊断及治疗过程,旨在提高对本病的认识,减少漏诊及探讨本病的治疗。
临床资料
一、一般资料
主诉:浮肿半年,血压高、尿检异常伴血肌酐高2周。
现病史:患者于2020年2月开始出现劳累后间断双下肢凹陷性浮肿,休息后缓解,未予诊治。至6月2日因浮肿加重于当地医院查血压160/100 mmHg,尿蛋白3+,尿蛋白定量 6.3g/24h,尿隐血3+,Alb 32g/L,血肌酐 152.1 umol/L,尿酸及甘油三酯升高高,ANA、A-dsDNA、抗核抗体谱、ANCA均阴性,Hb 95g/L,双肾超声提示弥漫性回声异常,双侧肾上腺超声未见异常,心电图、心脏超声未见异常,予降压、降脂、降尿酸、保肾对症治疗。6月9日我科门诊查尿蛋白定量3.90g/24h↑、尿沉渣红细胞计数978.9/μl↑、血肌酐1.52mg/dl↑、尿素氮22.20mg/dl↑、尿酸491μmol/L↑、白蛋白35.40g/L、血红蛋白105g/L↓、抗磷脂酶A2受体抗体1.77 RU/mL、ANCA阴性、抗GBM阴性,双肾超声示左/右126/127 mm,皮质回声增强。病程中患者无皮疹、关节痛、口腔溃疡、四肢麻木等表现。目前患者精神尚可,体力正常,食欲正常,睡眠正常,体重无明显变化,大便正常,排尿正常,为进一步检查及治疗“慢性肾炎”收治入院。
既往史、婚育史及个人史无特殊。
家族史:父亲年轻时出现浮肿,自述服用“偏方”治疗缓解,2019年复查尿检及肾功能正常,母亲体健,父母为近期结婚,为姑表关系,爷爷年轻时出现浮肿,服用“偏方”治疗缓解,未复查,3个姐姐体健,查尿检及肾功能均正常。
入院查体:体温:36.3℃,脉搏:78次/分,呼吸:16次/分,血压:160/100 mmHg,BMI 23.74 kg/m2,角膜混浊,全身无淋巴结肿大,双肺呼吸音清,未闻及干湿啰音;律齐,各瓣膜区未闻及病理性杂音;腹软,无压痛、反跳痛及肌紧张,移动性浊音阴性;双下肢未见浮肿。
二、检查
辅助检查:
(1)尿液检查:尿蛋白定量4.41g/24h↑、24小时尿量1670ml;尿常规示蛋白阳性(3+)、红细胞计数129.8/μl↑(混合性)、白细胞计数12.20/μl↑。尿NAG酶13.7U/g.cr↑、RB蛋白0.52mg/L、尿C3 2.00mg/L、尿a2-m 7.20mg/L↑、尿浓缩功能281mOsm/kg。尿本周蛋白阴性。
(2)血液检查:
血常规:白细胞计数4.07×109/L、血红蛋白88g/L↓、平均红细胞容积(MCV)100fl、平均红细胞血红蛋白量(MCH)32pg、平均血红蛋白浓度(MCHC)320g/L、网织红细胞百分比5.52% ↑ 、网织红绝对值152×109/L↑、血小板计数158×109/L、C反应蛋白<0.5mg/L。
血生化:白蛋白31.00g/L↓、球蛋白13.3g/L↓、肌酐1.51 mg/dl↑、尿素氮19.20 mg/dl、尿酸552 μmol/L↑、电解质未见明显异常、血清铁18.00 μmol/L、总铁结合力30.0 umol/L↓、不饱和铁结合力12.0 umol/L↓、铁蛋白183.4 ug/L、总二氧化碳27.70 mmol/L、前白蛋白247 mg/L、转氨酶及胆红素未见异常、总胆固醇(TCH)3.00 mmol/L、甘油三酯(TG)5.71 mmol/L↑、高密度脂蛋白胆固醇(HDL-C)0.26 mmol/L↓、低密度脂蛋白胆固醇(LDL-C)0.56 mmol/L、载脂蛋白AI0.46 g/L↓、载脂蛋白B0.62g/L、载脂蛋白E 64.5 mg/L↑、脂蛋白(a)21 mg/L、葡萄糖5.45 mmol/L、eGFR(CKD-EPI)62ml/min/1.73m2↓。HbA1c 3.7%。
(3)免疫:自身抗体:ANA、A-dsDNA、ENA抗体谱及磷脂抗体等均阴性,免疫球蛋白冷沉淀(IgG、IgM)21.82mg/L。补体C3、C4 未见降低,补体相关因子:H因子、I因子及H因子抗体等检查未见异常。直接抗人球蛋白(Coombs)及间接抗人球蛋白试验均阴性,外周血红细胞碎片未见。
肿瘤标志物及肿瘤相关抗原阴性。免疫固定电泳未见单克隆免疫球蛋白条带。血清游离轻链κ18.1mg/L、λ 23.2mg/L、比值0.78。
(4)骨髓检查:骨髓细胞学+流式检查可见增生性贫血,未见单克隆浆细胞。
(5)影像学检查及其他:大便常规+隐血 阴性;头颅骨盆平片未见明显X线异常。
消化道超声示脾大。脾脏:脾门厚45mm,肋下长39mm,全长149mm。脾内回声均匀。肝胆胰声像图未见占位。门静脉彩色多普勒检查未见异常。
眼科检查角膜混浊,眼底、眼压、视力均未见异常。
耳鼻喉科会诊纯音测听未见异常、扁桃体未见肿大。
基因检测示卵磷脂胆固醇酰基转移酶(LCAT)基因有1个纯合突变,在 1013 号核苷酸由胸腺嘧啶 T 变为胞嘧啶 C(c.1013T>C)的纯合突变,导致第 338 号氨基酸由亮氨酸变为脯氨酸(p.L338P),经家系验证分析,c.1013T>C(p.L338P)受检人之父该位点杂合变异,受检人之母该位点杂合变异。
(5)肾活检病理检查:免疫病理荧光IgM+、C3++、C1q+,弥漫分布,呈颗粒状沉积于系膜区、血管袢及袢内容物。IgG、IgA阴性。轻链染色未见明显异常。脂质荧光染色ApoB++、ApoE++,弥漫分布,呈颗粒状沉积于系膜区和血管袢。ApoB、ApoE袢腔阳性。ApoA阴性。
光镜:14个肾小球中1个球性废弃,1个节段硬化。余正切肾小球体积增大,系膜区中-重度增宽,系膜区见PAS阳性物质沉积,数处系膜溶解、节段毛细血管袢融合,袢腔内亦见PAS阳性物质,囊壁节段增厚。PASM-Masson:肾小球系膜区较多、内皮下、袢腔内节段、上皮侧少量嗜复红物沉积,节段外周袢分层,基膜可见空泡化改变。肾小管间质轻度慢性病变合并轻度急性病变,灶性小管萎缩、基膜增厚,管腔内见蛋白管型,间质灶性单个核细胞浸润,纤维化+。动脉未见明确病变。
电镜超微结构示:肾小球系膜区增宽,多数系膜溶解、结构疏松,系膜区较多块状不均匀的电子质密物沉积,高倍镜下其内见弯曲紊乱排列或平行排列的微管状物,直径9~13nm。较多毛细血管袢基膜内皮下疏松、区域明显增宽,致袢腔狭窄,偶见新的基膜形成,节段袢腔内、基膜内皮下节段、上皮侧散在上述电子致密物沉积。肾小球系膜区、基膜膜内偶见无特殊结构的电子致密物沉积。肾小球基膜内及系膜区较多空泡化变改变,个别空泡内见嗜鋨性物质。肾小球足细胞足突弥漫融合。
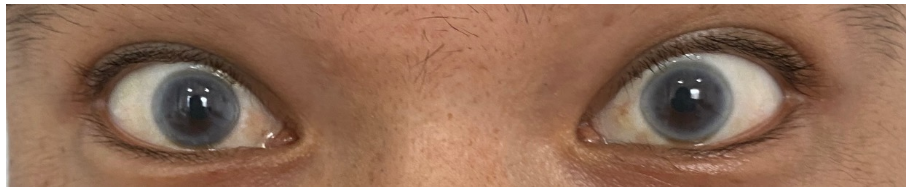
图1:患者眼角膜可见环状浑浊

图2.患者肾组织脂蛋白染色提示Apo B及Apo E弥漫阳性,沉积于系膜区,血管袢及袢腔内,而Apo A阴性。 A:Apo B阳性(免疫荧光×400) B:Apo E阳性(免疫荧光×400) C:Apo A阴性(免疫荧光×400)

图3:光镜下可见患者肾小球血管袢内PAS阳性物质沉积,外周袢分层 A:HE ×400B:PAS×400 C:Masson ×400 D:Masson+银染色 ×400
三、诊断与鉴别诊断
诊断:1.卵磷脂胆固醇酰基转移酶缺乏症
1.1卵磷脂胆固醇酰基转移酶缺乏症肾损害
本例患者临床表现包括大量蛋白尿,低蛋白血症,肾功能损伤,增生性贫血及角膜浑浊,脂质代谢异常,患者家族史中父亲及爷爷存在可疑家族史,基因检测提示基因纯合错义性突变,导致第 338 号氨基酸由亮氨酸变为脯氨酸,导致LCAT活性缺如,从而确诊卵磷脂胆固醇酰基转移酶缺乏症。
本例患者的鉴别诊断从以下几个方面考虑:
1.肾脏病变的鉴别:本例患者临床表现有大量蛋白尿,血压升高及肾功能损伤,排除禁忌证后行肾穿刺活检,免疫荧光检查提示肾组织IgG、IgA抗体均为阴性,可以排除临床常见的IgA肾病、膜性肾病等疾病;而患者肾组织IgM、C3等在系膜区、血管袢及袢内容物中均呈阳性,沉积形式并不常见,进一步进行脂蛋白染色提示脂蛋白ApoB、ApoE在肾小球中呈阳性,对比抗体及脂蛋白沉积分布特点,考虑免疫荧光IgM为非特异性染色,而查阅文献提示LCAT缺乏症患者既往有肾小球补体C3沉积的报道。患者电镜下超微结构检查提示肾小球血管襻内微管状物质沉积,进一步完善血液冷球蛋白、血液免疫固定电泳及游离轻链、骨髓穿刺流式细胞学等检查,排除了单克隆球蛋白相关疾病可能;电镜提示基膜内及系膜区较多空泡化变改变,个别空泡内见嗜鋨性物质,查阅文献既往曾有LCAT缺乏患者报道病理上可见肾小球基膜内病变,同时完善血清补体及补体相关因子等检查排除了可能导致肾小球基底膜内病变的电子致密物沉积病(Dense Deposit Disease,DDD)的可能,并由基因结果最终确认符合LCAT缺乏症相关肾脏损伤表现。

图4 电子显微镜显示患者超微结构 A:红色箭头示基底膜内空泡变性 B:黄色箭头示空泡变性内嗜鋨性物质
2.脂质异常的鉴别:肾病综合征患者由于大量蛋白尿,诱导肝脏脂质合成增加,常常导致胆固醇升高,其中尤其是低密度脂蛋白升高明显,但一般不会造成高密度脂蛋白的降低。本例患者高密度脂蛋白胆固醇仅0.26mmol/L,临床上以高密度脂蛋白胆固醇降低为主要表现的脂质异常疾病见下表:
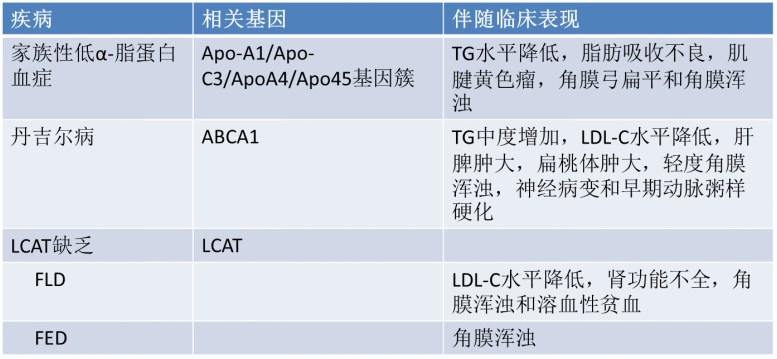
本例患者TG水平升高,不符合家族性低α-脂蛋白血症的表现,结合基因检测提示符合LCAT缺乏表现。同时患者为纯合基因突变,可导致LCAT活性缺失,结合临床表现除角膜浑浊外尚有尿蛋白、肾功能不全和溶血性贫血,这些表现不能被鱼眼病(FED)所解释,而符合家族性LCAT缺乏(FLD)的表现。
3.贫血的鉴别:本例患者临床表现为正细胞正色素性轻度贫血,铁代谢指标不符合缺铁性贫血表现,结合患者网织红细胞升高,骨髓穿刺结果等支持增生性贫血。询问病史及查体排除活动性失血可能,进一步完善Coombs试验等均阴性排除免疫性溶血可能。患者脾脏肿大,但血小板计数正常,亦不符合脾功能亢进表现,同时红细胞碎片阴性,不支持血栓性微血管可能。经查阅文献,提示LCAT缺乏患者由于成熟HDL的缺乏,会导致血浆内的脂蛋白与红细胞膜上的脂质交换出现障碍,引起过多的未酯化胆固醇在红细胞膜的蓄积,造成红细胞膜稳定性下降,已出现自发(非免疫性)溶血,造成溶血性贫血。同时亦有文献报道LCAT患者由于循环中未酯化胆固醇增加引起脾脏巨噬细胞大量吞噬,而氧化的脂质和脂质聚集物导致巨噬细胞上清道夫受体的激活和随后脂质的积累,促进泡沫状的组织细胞的形成。脂质在脾巨噬细胞中持续积累,导致泡沫状组织细胞增多,脾脏逐渐增大,最终脾肿大。
四、治疗
本例患者确诊后再治疗上予以双重血浆滤过治疗3次,总计置换及输注血浆3740ml、冷沉淀19.5u,并口服血管紧张素转换酶抑制剂贝那普利、降脂药物非诺贝特、阿昔莫司,并积极保肾、纠正贫血等药物治疗,出院前复查血色素升至103g/L,肌酐降至1.13mg/dl,白蛋白升至36.80g/L。
五、治疗结果、随访及转归
出院后1个月时随访时复查尿蛋白定量1.25g/24h↑、血红蛋白82g/L↓、体液免疫正常、学肌酐1.66mg/dl↑、尿素氮32.32mg/dl↑、尿酸563μmol/L↑、白蛋白41.50g/L、甘油三酯2.90mmol/L↑、高密度脂蛋白胆固醇0.17mmol/L↓,血肌酐回到治疗前水平,再次给予800ml新鲜冰冻血浆输注后患者血肌酐降低至1.55mg/dl,后随访血肌酐基本稳定。
讨 论
本例患者临床主要表现有浮肿伴蛋白尿,血肌酐升高,合并增生性贫血,这些症状在肾脏病患者中均为常见表现。但与多数肾脏病患者通过肾穿刺活检病理即可明确诊断有所不同,本例患者经肾脏病理检查并未能立即确诊。患者肾脏病理免疫荧光提示肾小球IgG、IgA均阴性,而IgM与C3的沉积部位也并不符合常见肾小球肾炎的表现,光镜下可见系膜区增宽伴系膜溶解,袢腔内异常物质沉积及外周袢分层,进一步通过电镜提示肾小球袢腔内物质的超微结构显示呈微管状,此类结构更常见于浆细胞病等血液病所导致的单克隆免疫球蛋白沉积,因此临床进行了多项相关检查(包括尿本周蛋白,血尿游离轻链、血清免疫蛋白固定电泳,球蛋白冷沉淀检测,骨髓穿刺流式细胞检测,肿瘤标记物、头颅骨盆平片寻找骨质破坏等)寻找血液疾病的线索,但无阳性发现;此外光镜所见的肾小球外周袢分层可见于膜增殖性肾小球病变,而在电镜下观察基底膜内存在空泡样改变,鉴于膜增殖性病变中的DDD可造成基底膜内病变,因此临床上也进行了补体检测以及一系列相关调节因子(如H因子、I因子及其抗体等)检查,亦未见异常,可排除DDD可能。
至此,我们再度回到临床寻找患者疾病的线索,注意到患者角膜出现特殊的环状浑浊以及血清HDL-C显著降低,追问病史发现患者父母为近亲结婚,考虑到患者存在LCAT缺乏的可能。于是病理上进一步完善脂蛋白染色,证实患者肾小球内ApoB及ApoE脂质均为强阳性,并进一步比较沉积部位与形式,综合考虑患者IgM为脂质沉积所导致的非特异性染色,而查阅文献报道LCAT患者肾组织C3沉积呈阳性,同时亦有文献报道提示LCAT患者肾小球基底膜可能出现空泡变性,并进一步完善患者相关基因检查后最终得以确诊。
LCAT缺乏症是一种非常罕见的常染色体隐性脂质代谢紊乱,其原因是LCAT基因功能的缺失。LCAT是人体内唯一能使胆固醇酯化的酶,在血管内HDL-C的重塑和HDL-C水平的稳定中起着核心作用。LCAT在巨噬细胞反向胆固醇运输中也起关键作用,该运输是将多余的胆固醇从外周细胞中排出并输送到肝脏排泄的过程。
血浆中的LCAT主要存在两种活性:(1)α-LCAT活性,针对HDL中胆固醇底物,需要ApoA1和ApoA4 作为其辅助因子;(2) β-LCAT 活性是专门针对LDL、VLDL 中的胆固醇,不需要辅助因子。基于其活性缺失的程度,LCAT缺乏症分为两类:1.家族性LCAT 缺乏症( FLD) :α-LCAT 和β-LCAT 活性均缺乏或降低,造成血浆中未酯化胆固醇升高,临床表现为角膜混浊、正色素性贫血及肾脏损伤。2.鱼眼病( FED) :α-LCAT 活性缺乏,而β-LCAT 活性不变,血浆中未酯化胆固醇正常或轻微上升,临床表现为角膜混浊而肾功能正常。
回顾本例患者,其临床符合典型FLD表现,父母为近亲结婚(姑表关系),进一步的基因验证提示其父母均为(c.1013T>C)的杂合突变,由于该病为常染色体隐性遗传,其父母均未发病,其兄弟姐妹亦无相关表现,但患者不幸继承父母双方的致病基因,成为(c.1013T>C)的纯合突变,继而发病,出现FLD相关表现,本例患者家族中其他成员尚未完善基因检测。
本例患者临床表现典型FLD,但由于LCAT缺乏症临床较为罕见,其诊断过程仍有一定曲折。本病目前尚无针对性的治疗方法,有文献报道血管紧张素转化抑制剂及调脂治疗部分改善患者预后。鉴于本病起因为基因异常导致的LCAT缺乏,本例患者尝试采用血浆置换的方式大量补充健康人的新鲜血浆,以期弥补患者体内缺失的LCAT活性,观察显示患者短期内血肌酐及贫血、尿蛋白均有改善,但在后期的随访复查过程中患者肌酐逐渐回到治疗前水平,提示大量补充新鲜血浆能在短期内改善患者症状,但持续时间较短。重组LCAT补充疗法可能是今后临床改善该疾病预后的希望所在。
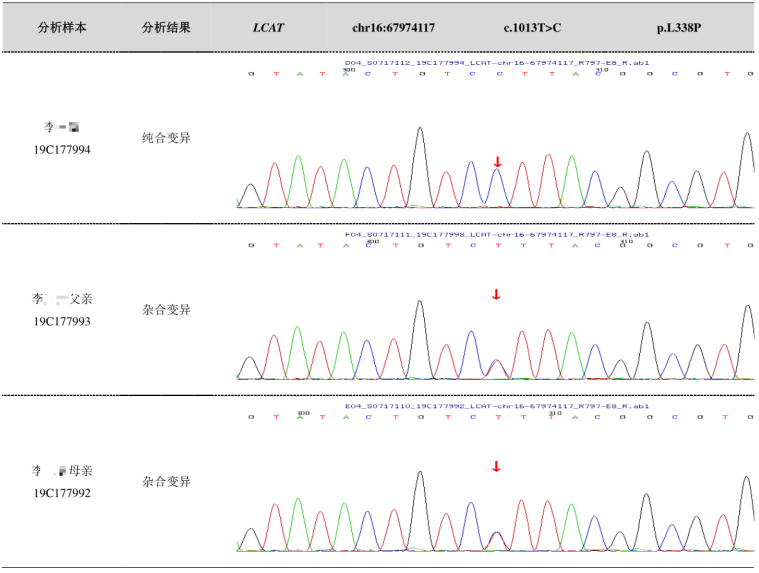
图5:患者及其父母基因检测结果
参考文献:略
来源:东部战区总医院供稿
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017