




200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者 北京天坛医院 蒋梦雪 张亚清 王永刚
偏头痛是一种最常见的慢性神经血管相关的原发性头痛,以单侧、发作性和搏动样头痛为主要表现,可伴有恶心、呕吐等症状,一般持续4~72 h,光、声刺激或日常活动均可加重头痛,安静环境或休息后头痛可缓解。全球约10.4亿人患有偏头痛,男性终身患病率约10%,女性约22%[1-2]。我国偏头痛的年患病率为9%,确诊为偏头痛的患者每年治疗成本超过2994亿元[3-4]。
近年来,针对其发病机制的研究持续进行,但仍尚不明确。目前认可度较高的发病机制是三叉神经血管障碍学说和皮层扩散抑制(CSD)学说,中枢敏化、降钙素基因相关肽(CGRP)、神经胶质细胞等因素在其中起着重要作用[5]。目前,治疗偏头痛的方法仍存在不足,进一步探索其发病背后的病理生理学机制对有效的治疗尤为重要。本文将简述偏头痛的相关病理生理学机制及最新发现。
一、偏头痛的发病机制及相关标志物
1. 三叉神经血管障碍与中枢敏化机制
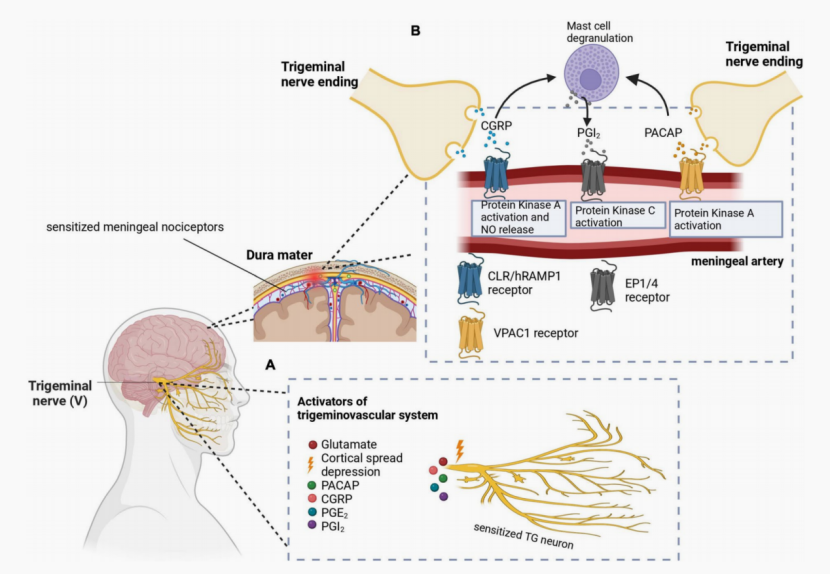
基于三叉神经血管障碍理论,偏头痛的核心病理过程与三叉神经血管系统(TGVS)的敏化密切相关(见图1)。在多种触发因素(如应激、炎症等)作用下,释放的促炎因子导致中枢痛觉信号处理阈值降低,中枢兴奋性与抑制性平衡发生紊乱,使得TGVS处于敏化状态,进而引发脑膜无菌性神经源性炎症、肥大细胞脱颗粒和脑膜血管舒张等一系列反应[5]。同时,敏化的三叉神经末梢释放CGRP和P物质(SP)等神经肽。CGRP作为一种强效扩血管的神经肽,在中枢敏化及偏头痛发作中起关键作用:可直接引起血管扩张,激活神经胶质细胞,通过正反馈维持炎症和神经高敏状态[6]。神经肽含量的增加可促进白细胞介素(IL)-1β、IL-6、肿瘤坏死因子(TNF)-α等炎症因子以及一氧化氮(NO)、氧自由基等的释放,从而诱导偏头痛发生[7]。
图1 三叉神经血管系统参与偏头痛发病的分子与病理机制示意图
2. CSD与先兆偏头痛
此前一些单基因综合征的研究发现,CSD是与有先兆偏头痛密切相关的机制。其核心过程为:CSD在大脑皮层起始后缓慢扩散,通过触发神经元和小胶质细胞去极化而激活TGVS,从而参与头痛发作[5]。星形胶质细胞的非钙离子依赖性信号通路和小胶质细胞电压敏感离子通道的激活均参与CSD的启动和传播,与视觉、感觉等先兆症状直接相关[8-9]。同时,谷氨酸的活性增加也被认为是CSD产生的关键[10]。
3. 神经胶质细胞的活化与调控
在偏头痛发病时,星形胶质细胞通过摄取利用γ-氨基丁酸(GABA)和谷氨酸,生成谷氨酰胺调节神经递质平衡;在中枢神经系统中,其释放的S100β蛋白在高浓度时可刺激炎症因子表达,进而参与三叉神经致敏;同时,三叉神经节中的星形胶质细胞可释放炎性因子维持痛觉敏感状态[5]。小胶质细胞作为中枢免疫细胞,活化后通过Toll样受体(TLR2、TLR3、TLR4)、嘌呤受体(P2X4、P2X7、P2Y12)等激活,经脑源性神经营养因子(BDNF)/原肌球蛋白相关激酶B(TrkB)、NOD样受体家族含pyrin结构域(NLRP)3/IL-1β等通路释放IL-1β、IL-6、TNF-α等细胞因子,增强周围神经元的兴奋性,从而促进中枢敏化和慢性偏头痛进展[11]。
4. 细胞信号通路的参与
关键信号通路在偏头痛的触发、维持和加重中起重要作用。丝裂原活化蛋白激酶通路由c-Jun氨基末端激酶(JNK)、p38丝裂原活化蛋白激酶和细胞外信号调节激酶(ERK)组成,可被TNF-α、环氧合酶(COX)-2等上游信号激活,从而调控炎症反应、氧化应激和突触可塑性等,激活后通过下游的c-Fos和c-Jun(早期即刻基因)参与痛觉调控,CGRP则可通过激活ERK通路进一步活化转录因子反应结合蛋白(CREB)——慢性偏头痛大鼠的中枢致敏指标。核因子-κB(NF-κB)通路通过上调一氧化氮合酶(NOS)、COX-2等炎症相关酶的转录,从而调节炎症因子表达,同时调控胶质细胞增生和免疫反应来参与偏头痛的发病[5]。
5. 垂体腺苷酸环化酶激活肽(PACAP)
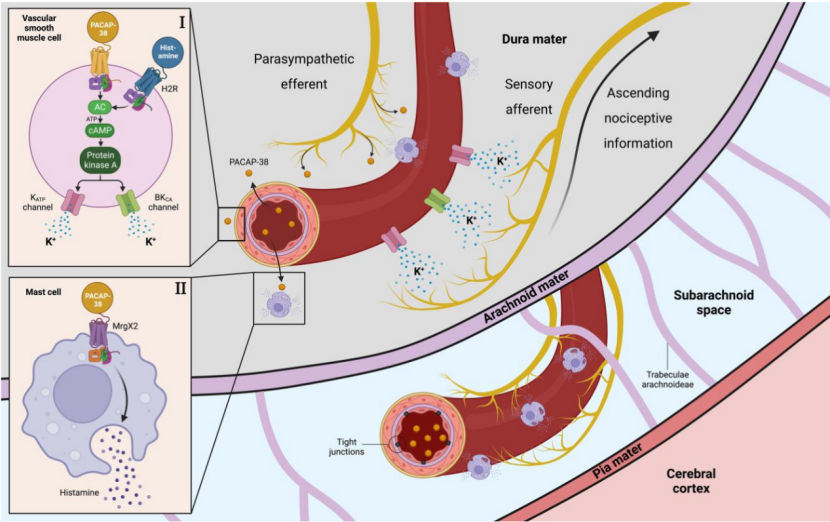
PACAP作为独立于CGRP的神经肽,在哺乳动物中以PACAP38为主要形式,通过G蛋白偶联受体(如PACAP1R)介导颅内动脉扩张或肥大细胞脱颗粒来激活脑膜痛觉感受器,从而诱导偏头痛样头痛发作(见图2)。
图2 垂体腺苷酸环化酶激活肽-38 介导的偏头痛脑膜神经血管交互作用机制图
血管标志物内皮素(ET)(尤其是ET-1)作为强缩血管物质,可能通过与三叉神经节中的受体(ET-A、ET-B)结合,参与CGRP诱导的疼痛反应;IL-1β、IL-6、TNF-α和高敏C反应蛋白等炎症因子通过促进神经源性炎症和中枢敏化来参与偏头痛的启动和维持[11]。
1.脑脊液流动机制
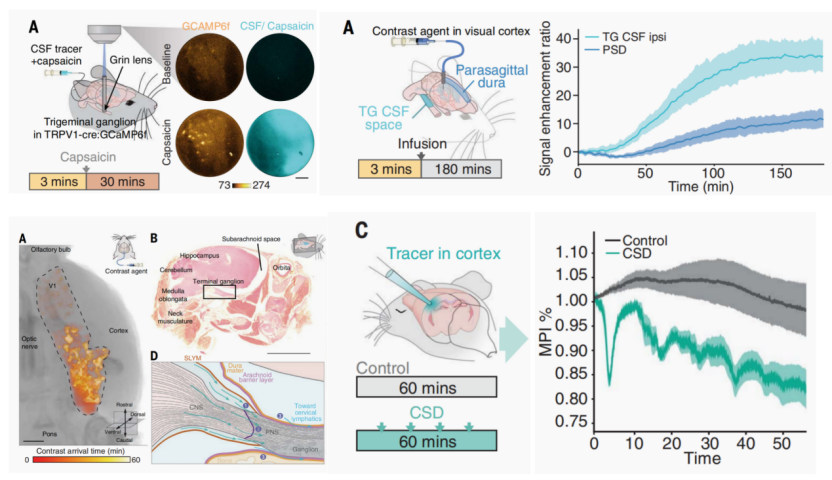
根据2024年发表于《科学》(Science)上的研究发现,在偏头痛模型中,脑脊液(CSF)可直接流入三叉神经节,建立中枢神经系统与外周神经系统的非突触信号通路(见图3)。此外,研究表明CSD会改变约11%的CSF蛋白质组,上调包括CGRP在内的七种蛋白质水平,从而促进视觉皮层的细胞外溶质向三叉神经节流动,激活三叉神经节神经元活动[12]。这一研究结果解释了偏头痛先兆与头痛发作的关联,为偏头痛发病机制提供了新的结构和分子基础。
图3 皮层扩散抑制诱导脑脊液分子变化并流入激活三叉神经节
2. 基因突变
相关研究发现,酪蛋白激酶1δ(CK1δ)基因突变会引起突触后神经重塑,导致突触前适应缺陷和谷氨酸释放增加,从而显著增加偏头痛的易感性。
3. 阈值假说
根据2024年发表的一项针对爱丽丝梦游仙境综合征(AIWS)的综述[13],偏头痛患者存在皮层兴奋性升高及习惯化缺陷,这本质上反映了大脑对刺激的反应阈值降低。而AIWS作为一种与偏头痛相关的特殊先兆症状,恰恰间接支持了偏头痛的阈值假说,即偏头痛患者存在皮层兴奋性阈值降低,使得大脑对内外刺激的敏感性增加,导致神经系统出现异常神经活动,更容易触发偏头痛发作。
目前最成熟的偏头痛治疗靶点聚焦在CGRP及其受体,已经实现临床转化,的新型药物包括CGRP受体拮抗剂(如Atogepant、Ubrogepant和Rimegepant)、CGRP单克隆抗体(如Erenumab、Eptinezumab、Galcanezumab)以及5-羟色胺(5-HT)1F受体激动剂(如Lasmiditan)[14]。
近年来,偏头痛治疗的研究热点集中在PACAP及PAC1受体。在一项2期临床试验中,PAC1受体拮抗剂取得了阳性结果,在未来有潜力弥补CGRP靶点的局限性并拓宽治疗覆盖人群。除了这一靶点,小分子肽以及各种三叉神经节离子通道等也进入临床试验阶段。
综上所述,偏头痛的发病机制涉及多方面,不同机制之间存在交互。尽管针对偏头痛相关发病机制的研究在不断取得新的进展,但目前仍不能够彻底明确,这就导致在偏头痛对因治疗方面依旧存在较大局限,许多患者对当前治疗靶点的疗效不佳,导致偏头痛反复发作,甚至严重影响患者的社交及生活质量。
所以,目前亟须明确偏头痛的发病机制,在未来可以实现针对不同患者的不同发病机制来采用针对性的治疗手段,帮助患者从根源上治疗疾病,提升患者的生活质量和幸福程度。
Take Home Message
1.三叉神经血管障碍与中枢敏化、CSD、神经胶质细胞与信号通路、其他因素(如PACAP独立于CGRP通路、血管内皮素和炎症因子)均为目前研究显示的偏头痛相关发病机制。
2.最新研究结果怀疑CSF流动机制、阈值假说及基因突变也可增加偏头痛易感性,从而引发偏头痛发作。
3.基于当前研究,CGRP及其受体已经成功进行临床转化并作为治疗偏头痛的成熟靶点,而PACAP被证实为独立于CGRP通路的潜在靶点,有望在将来弥补CGRP靶点治疗的局限性,提高偏头痛的治愈率。
专家简介

A. 促炎因子释放降低痛觉信号处理阈值
B. 星形胶质细胞通过非钙离子依赖性信号通路激活CSD
C. 三叉神经末梢释放CGRP引发血管舒张
D. 炎症因子(如IL-1β、TNF-α)促进神经源性炎症
02 (多选题)关于偏头痛的最新研究进展,下列说法正确的有哪些?
A. CSD可改变脑脊液蛋白质组,上调CGRP等蛋白质
B. CK1δ基因突变通过增加谷氨酸释放提高偏头痛易感性
C. 阈值假说认为偏头痛患者皮层对刺激的反应阈值升高
D. 脑脊液可直接流入三叉神经节,建立中枢与外周的信号联系
(评论区留言一起讨论一下吧~)
Day8 答案明日揭晓

点击图片查看更多精彩内容
最终解释权归中国医学论坛报社所有,转载须授权
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017